A Four-Year-Old Boy with a Mass in the Left Leg
February 5, 2014
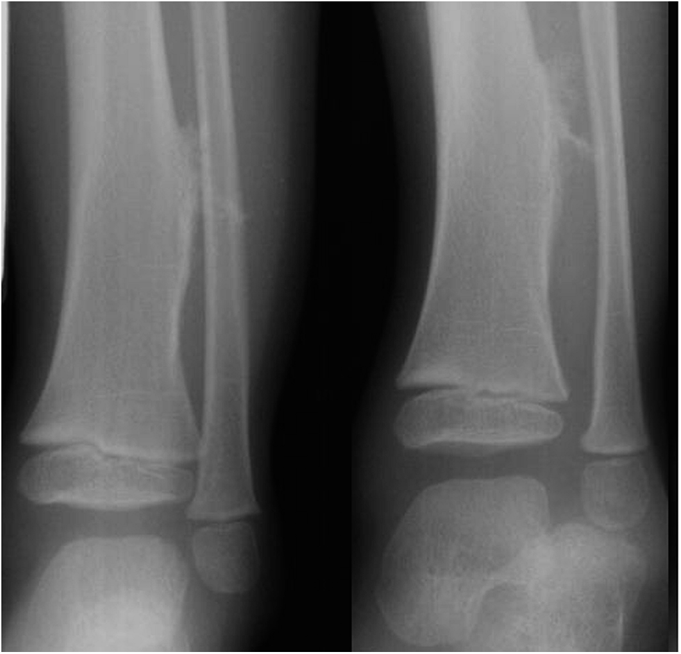
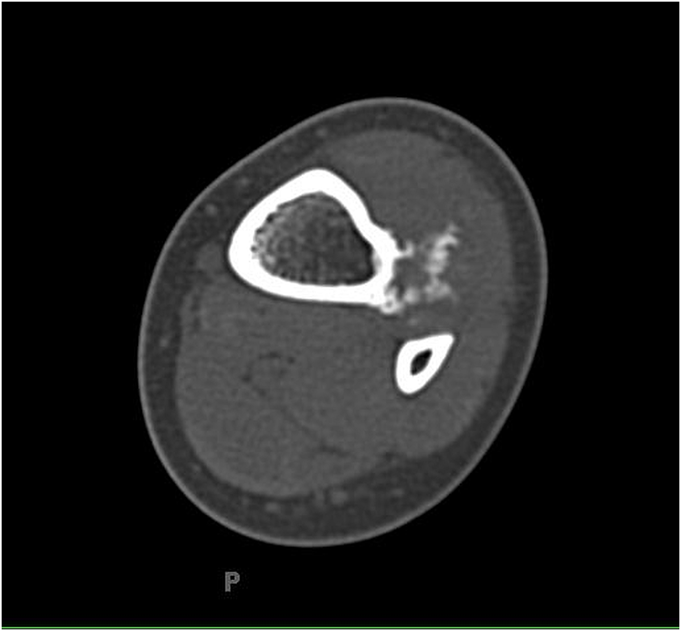
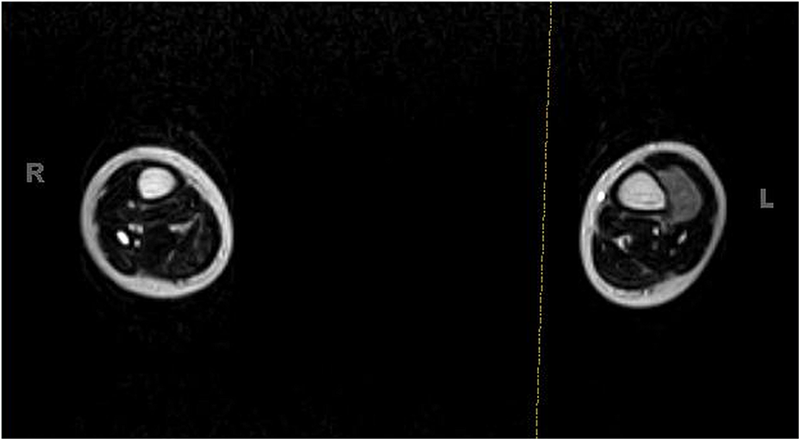
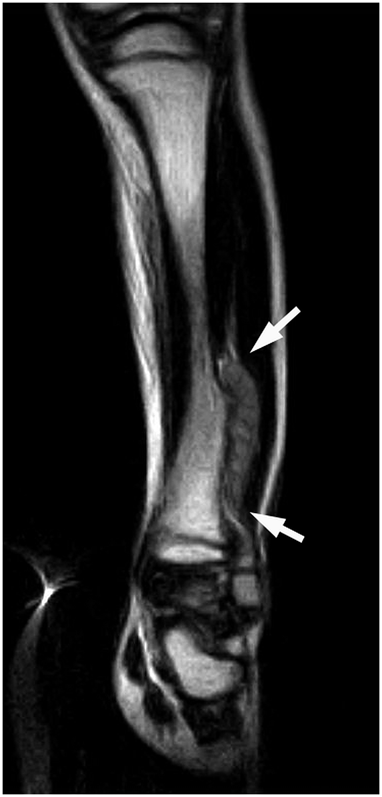
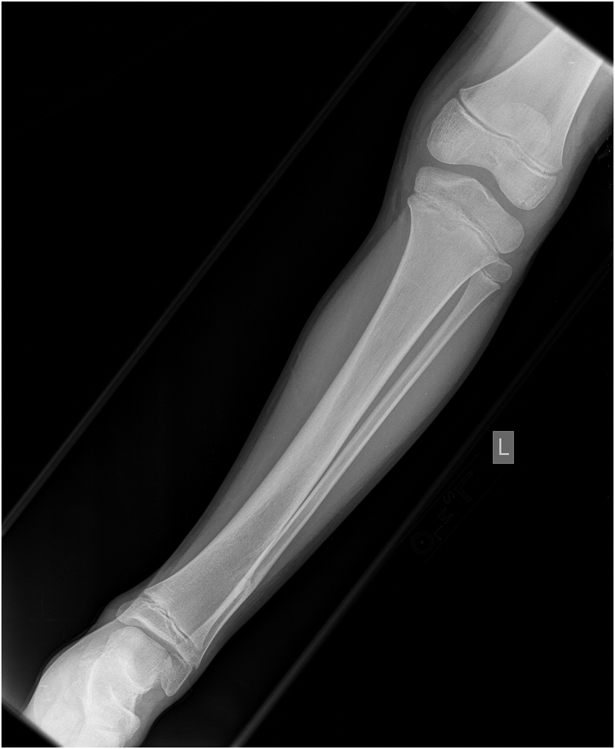
 Fig. 1
Fig. 1 Fig. 2
Fig. 2 Fig. 3-A
Fig. 3-A Fig. 3-B
Fig. 3-B Fig. 4
Fig. 4 Fig. 5
Fig. 5 Fig. 6
Fig. 6 Fig. 7-A
Fig. 7-A Fig. 7-B
Fig. 7-B Fig. 8
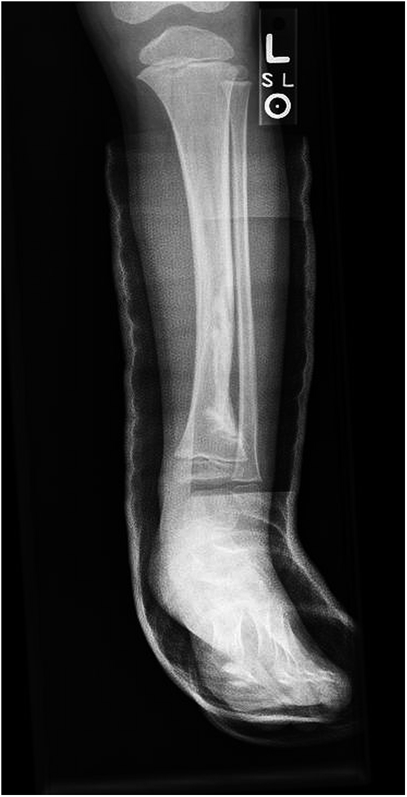
Fig. 8A previously healthy four-year-old boy presented to his primary care physician after his grandmother noticed a mass in the left leg above the ankle. The patient was asymptomatic and had an unremarkable medical history. Physical examination revealed strong distal pulses, no sensory or motor deficits, and a firm, painless soft-tissue mass on the lateral aspect of the distal part of the left leg. Radiographs of the distal part of the left tibia and fibula showed spiculated calcifications arising from the lateral cortex of the distal part of the tibia and minimal periosteal reaction (Fig. 1). Although magnetic resonance imaging (MRI) with contrast would have been the next preferred imaging study because of its superior soft-tissue characterization and lack of ionizing radiation, the primary care physician obtained a computed tomography (CT) scan to evaluate for osteochondroma. CT without contrast demonstrated a mass arising from the distal part of the left tibial cortex with some areas of mineralization (Fig. 2). Although osteochondroma was part of the differential diagnosis, a lack of complete corticomedullary continuity raised the suspicion of a periosteal osteosarcoma; thus, an MRI scan was obtained. This scan showed a somewhat heterogeneous mass adjacent to and infiltrating the periosteum of the lateral aspect of the distal part of the left tibia, with an associated soft-tissue mass extending into the surrounding muscle (Figs. 3-A and 3-B). The patient was then referred to our orthopaedic oncology department for definitive care. Subsequently, a core needle biopsy was obtained.
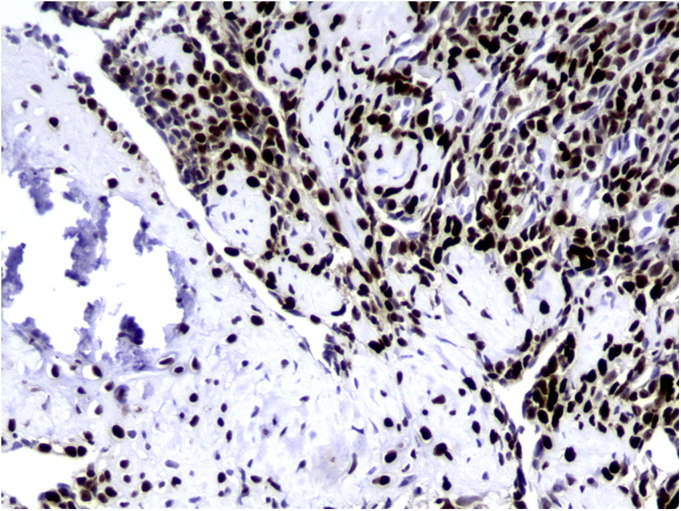
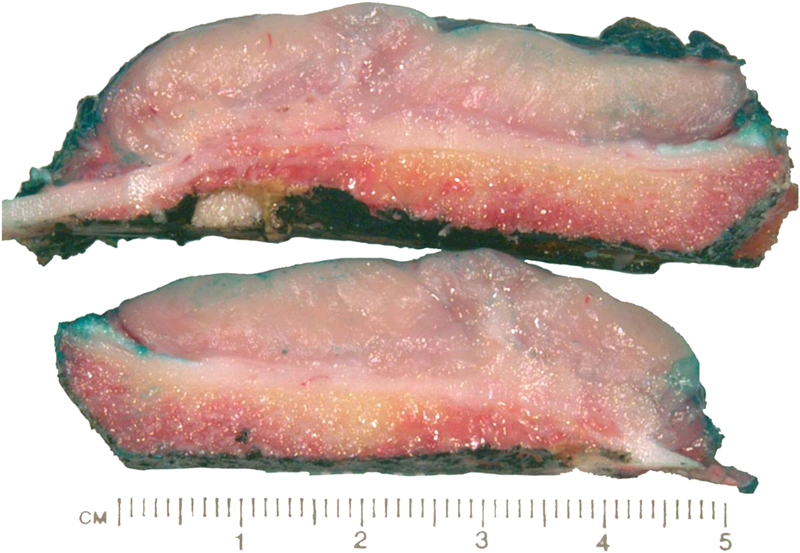
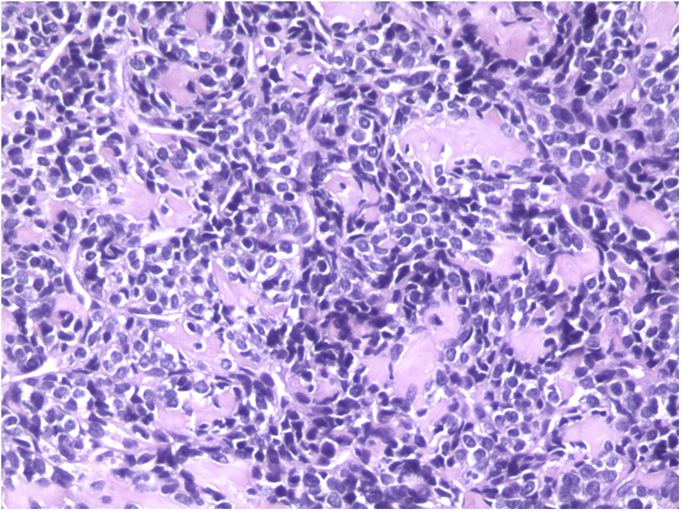
The tumor cells were positive for the CD99 marker (membranous) and focally for the S100 protein. The tumor cells were negative for other markers, including keratin, epithelial membrane antigen, HMB45, synaptophysin, PGP9.5, desmin, and CD45. The diagnosis of mesenchymal chondrosarcoma was additionally supported by strong staining for Sox9 by immunohistochemistry (Fig. 4) and pertinent documentation of an absent Ewing sarcoma breakpoint region or SYT gene rearrangement by fluorescent in situ hybridization on paraffin sections. Staging was completed with a CT scan of the chest, which showed no evidence of pulmonary metastasis. The patient received four cycles of neoadjuvant chemotherapy according to the AEWS1031 protocol, with no progression or regression of tumor size on physical examination. He then underwent hemicortical resection of the midshaft of the left tibia, extending distally to within 1 cm of the epiphysis. The resected area was packed with powdered allograft bone, and primary closure was attained without complication (Fig. 5). He was discharged home with a short leg cast on postoperative day four. Final pathology evaluation demonstrated a 5.4 × 2.5 × 1.0-cm firm, fibrous, lobulated mass arising from the outer cortex and periosteum of the tibia (Fig. 6). There was focal disruption of cortical bone in the proximal third of the lesion. The surgical margins were uninvolved. There were focal areas with inconclusive osteoid matrix formation (osteoid-like material without calcification). The mitotic index was 2/10 high-powered fields. The proliferative index averaged 4%. The tumor was composed of primitive undifferentiated cells growing diffusely amid collagenized tissue, but there was no conclusive osteoid matrix formation by the tumor. In some areas, the tumor exhibited a hemangiopericytic pattern with focal differentiation into low-grade mature hyaline cartilage (Figs. 7-A and 7-B). The specimen showed a grade-II response to chemotherapy with tumor necrosis of 50% to 90%; there was no confluent necrosis. Because of the minimal response to chemotherapy and the complete excision of the mass, the decision was made to forgo any postoperative chemotherapy or radiation. The patient had no complications postoperatively, and he advanced to full activity after cast removal four weeks postoperatively. After more than 4.5 years of regular follow-up, including clinical examination, restaging CT of the chest, radiographs of the left tibia (Fig. 8), and MRI of the left leg, there was no sign of recurrent or metastatic disease. The patient was playing sports year-round without restrictions or residual symptoms.
Proceed to Discussion >>Reference: Nugent D, Cheon D, Montforte H, Ayala AG, Letson D, Keen J. Periosteal mesenchymal chondrosarcoma in the distal part of the tibia of a four-year-old boy: a case report. JBJS Case Connector, 2013 Nov 27;3(4):e117.
The patient’s age, the site in the distal part of the tibia, and the periosteal location of the tumor in this case make it an extremely unusual presentation of a rare disease. A thorough search of the literature uncovered only three cases of periosteal mesenchymal chondrosarcoma, all of which were in adult femora. Only a handful of cases in children under the age of ten were found, none of which were located in the extremities. To the best of our knowledge, this is the first case of periosteal mesenchymal chondrosarcoma reported in a child and that he is the youngest reported patient with this malignancy occurring in an extremity. The diagnosis of mesenchymal chondrosarcoma can be confidently made by light microscopy in most instances by its distinctive primitive cell morphology, its growth pattern, and the usual presence of low-grade hyaline cartilage islands that may undergo endochondral ossification. The age of our patient, the rarity of the topography of the chondrosarcoma, and some of the histologic findings warranted exclusion of alternative diagnoses, including poorly differentiated synovial sarcoma and Ewing sarcoma. Additionally, the morphologic absence of conclusive osteoid matrix amid the tumor cells, both in the biopsy or postchemotherapy resection specimen, and the strong Sox9 nuclear positivity further confirm the cartilaginous lineage of this tumor and confidently exclude osteosarcoma, as previously reported by Wehrli et al. in 2003 and more recently by Fanburg-Smith et al. in 2010. A recent review of the utility of Sox9 highlights the potential overlap of its expression in other neoplasms, particularly chondroblastic components of osteosarcoma and synovial sarcoma, reminding us of the importance of integrating morphologic findings with esoteric testing when warranted. Mesenchymal chondrosarcoma typically has an aggressive course that is characterized by local recurrence and/or distant metastasis. Historically, the prognosis has been dismal; some reports have described the disease as "highly lethal" and "almost uniformly fatal." Indeed, many studies have found the ten-year survival rate to be less than 50%, and one report described an 80% mortality rate after ten years in a pediatric population. However, Dantonello et al. reported an overall ten-year survival rate greater than 60% in a series of fifteen children with mesenchymal chondrosarcoma, which may either be a result of patient selection or a sign of more effective therapy. Additional studies are needed to form consensus guidelines for the management of this unusual tumor. Currently, complete surgical excision with the addition of chemotherapy and/or radiation is the treatment of choice; the use of chemotherapy and radiation therapy must be better characterized. As more outcomes are reported, it will be important to define whether mesenchymal chondrosarcoma in children and adolescents responds, and is therefore treated, differently than in older populations, as proposed by Dantonello et al. Additionally, as proposed by Huvos et al., medical therapies may be developed or refined to target specific histologic subtypes. Finally, the scarcity of reported cases, especially over the last two decades, accentuates the need for multicenter studies to determine the most appropriate management of this rare malignancy.
Reference: Nugent D, Cheon D, Montforte H, Ayala AG, Letson D, Keen J. Periosteal mesenchymal chondrosarcoma in the distal part of the tibia of a four-year-old boy: a case report. JBJS Case Connector, 2013 Nov 27;3(4):e117.

Parosteal osteogenic sarcoma
Osteochondroma
Periosteal mesenchymal chondrosarcoma
Intracortical chondrosarcoma
Ewing sarcoma