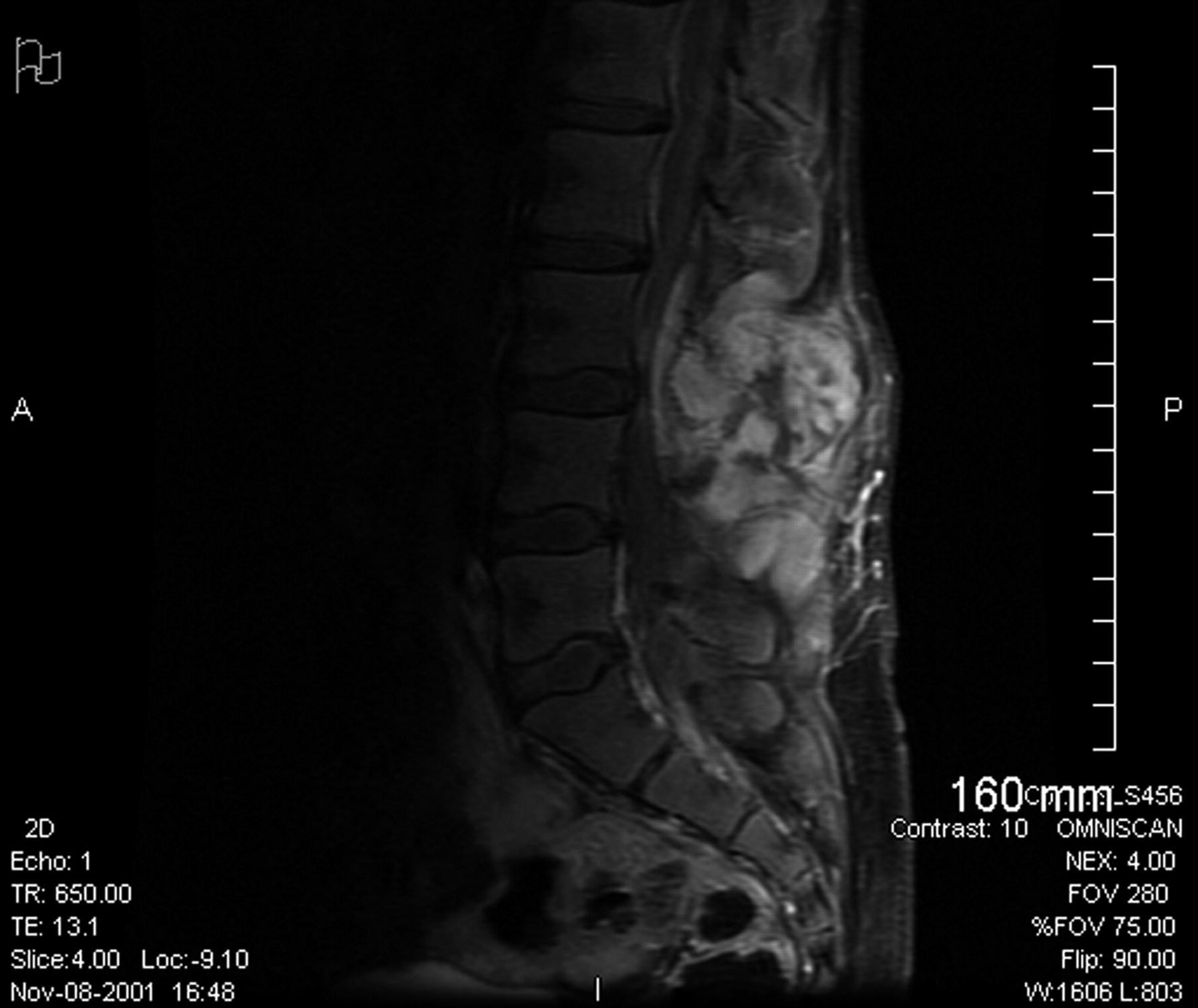
 Fig. 1
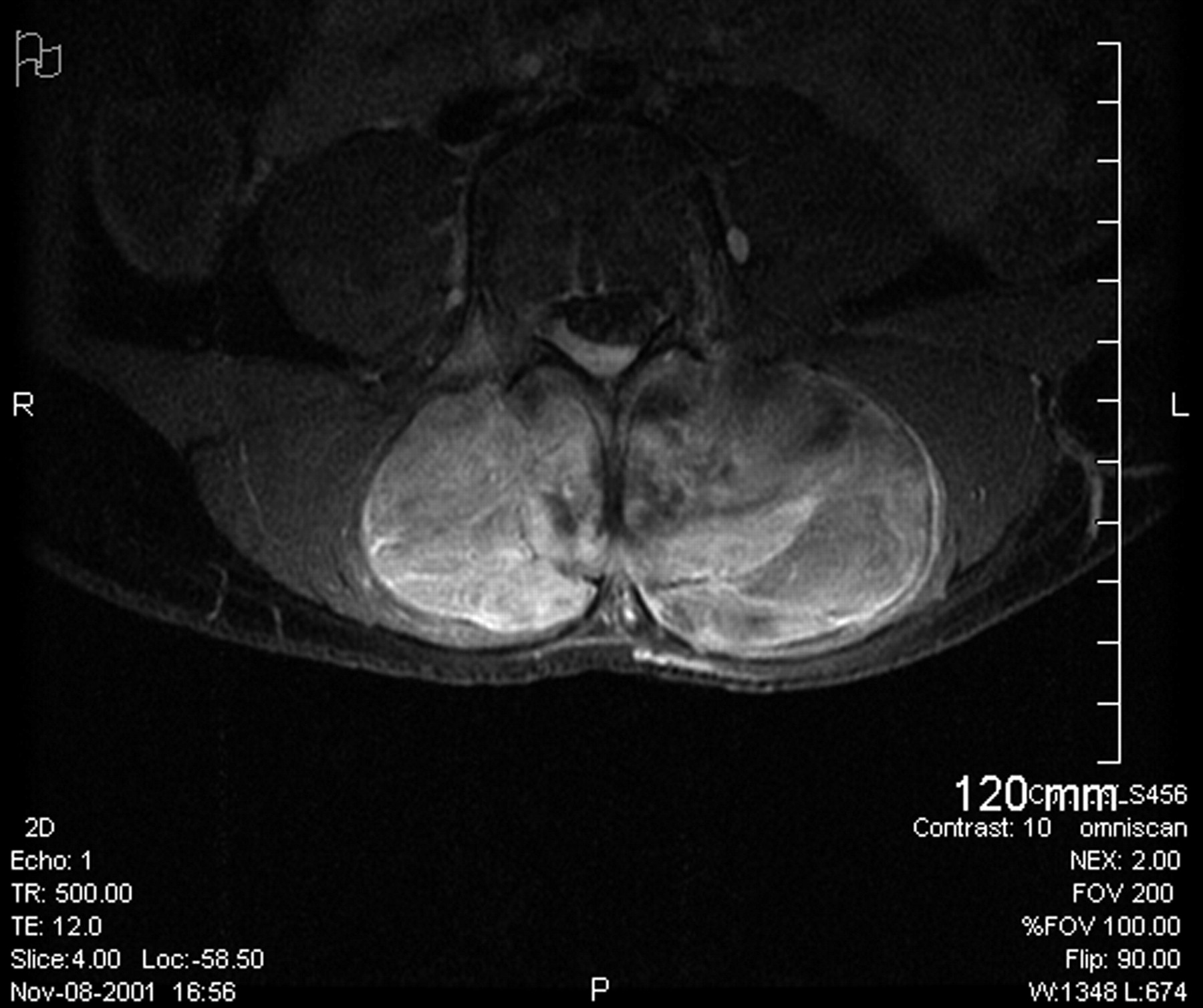
Fig. 1 Fig. 2
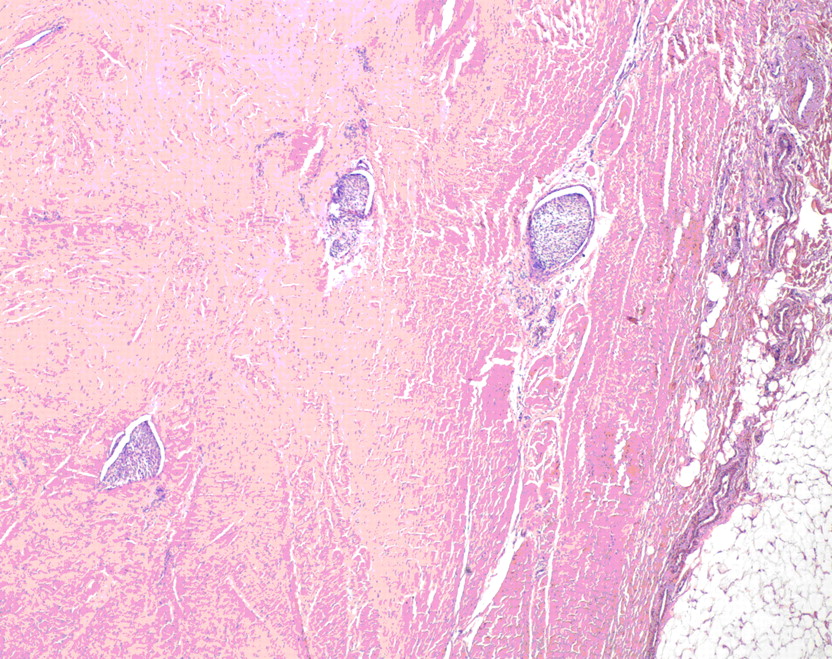
Fig. 2 Fig. 3
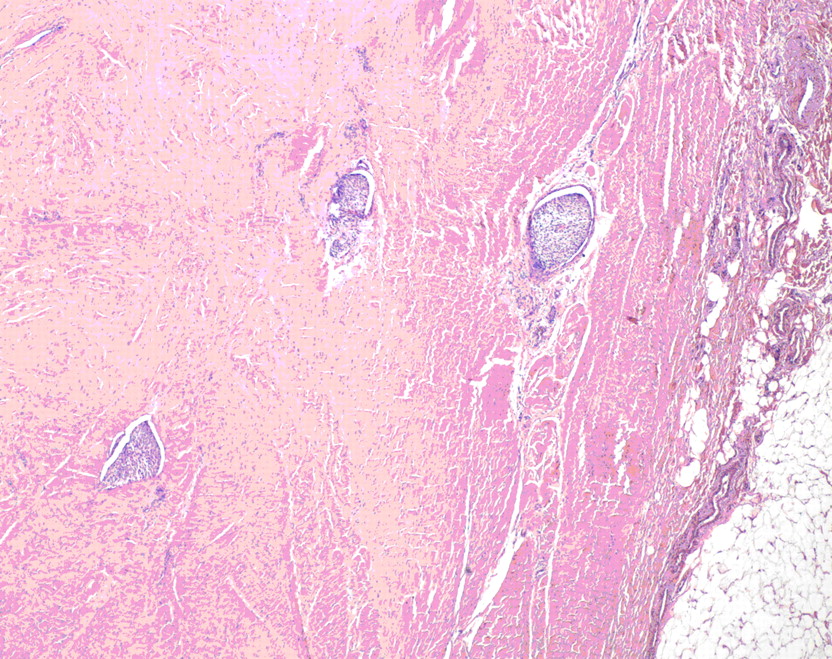
Fig. 3 Fig. 3
Fig. 3 Fig. 4
Fig. 4 Fig. 5
Fig. 5A fourteen-year-old girl with no related medical history presented to her pediatrician in November 2001 with unresolved lower back pain two weeks after being struck in the lumbar region while playing soccer. Following this incident, she began experiencing paresthesias in the left lower extremity (and occasionally in the right lower extremity) and on questioning was able to recall a history of mild and occasional low-back pain during the previous two months. Physical examination revealed a firm, fixed palpable mass in the lumbar paraspinal muscles, and the patient was referred for imaging studies. No neurologic examination was noted at that time. A magnetic resonance imaging scan of limited quality secondary to patient movement showed a large lesion that was located mostly in the lumbar paraspinal muscles with extension into the spinal canal at the L3-L4 level with resultant compression of the thecal sac. The patient was referred for orthopaedic consultation and was transferred to our regional academic health-care center. Physical examination revealed a large, firm, fixed mass in the lumbar region bilaterally, extending from approximately L2 to L5. The review of systems was negative for weight loss, fevers, night sweats, bowel or bladder dysfunction, signs of systemic illness, or impairments of gait or balance. There were no overlying skin changes, and the mass was not tender to palpation. No adenopathy was noted in the cervical, supraclavicular, axillary, or inguinal regions. Muscle strength testing was normal on the right side but revealed 4/5 strength for the extensor hallucis longus, tibialis anterior, peroneal, and gastrocnemius muscles on the left side, with no deficits of sensation. Examination of the reflexes revealed a negative ankle jerk bilaterally and a 2+ right-sided patellar response, although the left-sided patellar reflex was absent. Perineal sensation was intact, and rectal tone was normal. A repeat magnetic resonance imaging scan, performed with intravenous contrast medium, showed a 9.4 × 5.4 × 10.8-cm soft-tissue mass extending from L2 to the sacrum with osseous invasion of the L3 and L4 pedicles (Figs. 1 and 2). Osseous erosion of the L3, L4, and L5 posterior elements with extension of the tumor into the spinal canal with resultant extrinsic compression of the neural elements also was noted. A total-body planar bone scan showed decreased uptake in the posterior elements of L2, L3, and L4. There was no soft-tissue uptake. A chest, abdomen, and pelvic computed tomography scan (performed to look for adenopathy or other sites of disease) and laboratory studies showed no evidence of metastatic disease or adenopathy. An open midline posterior incisional biopsy at L2 was performed (Fig. 3).
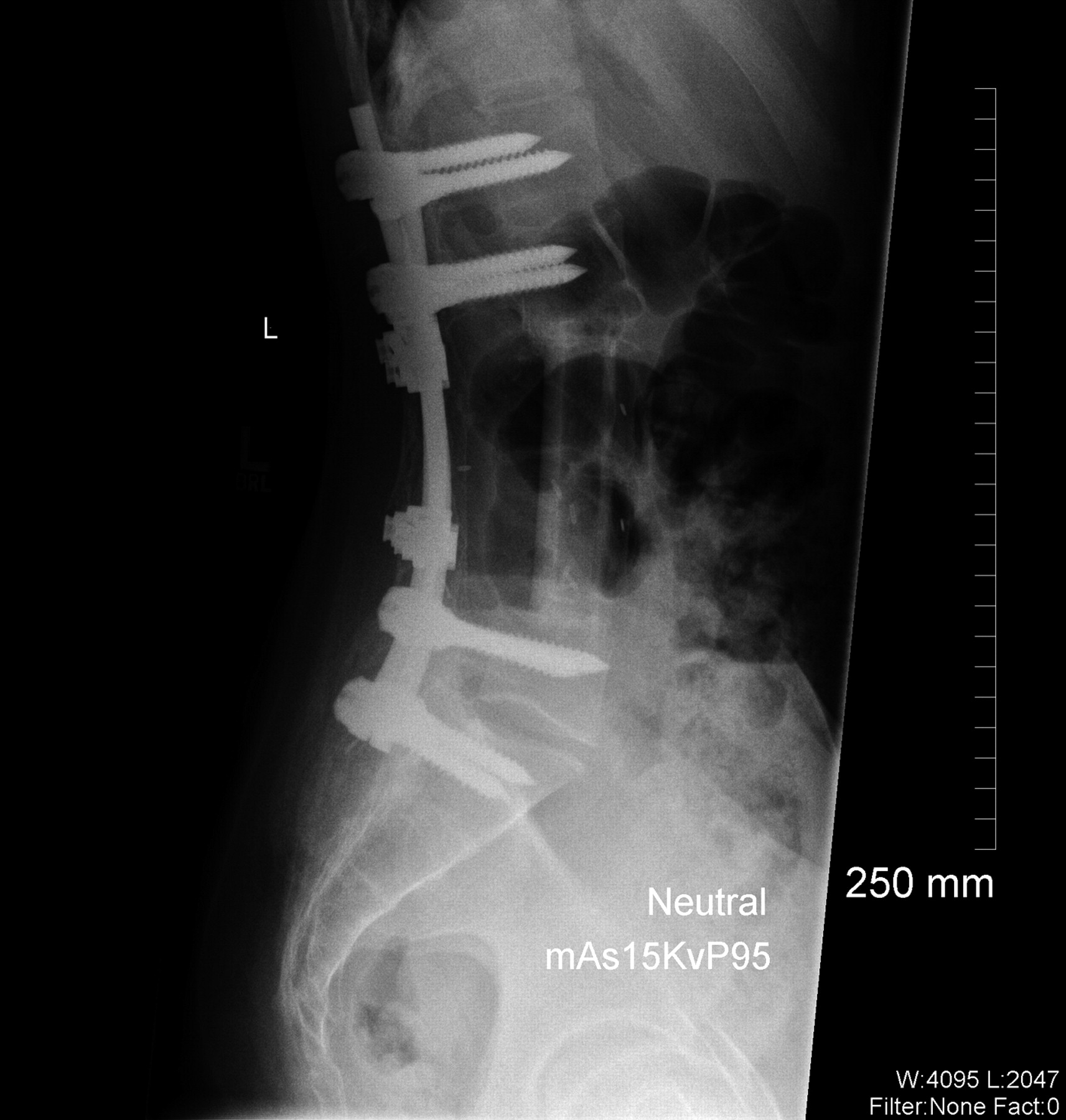
A frozen-section and subsequent cytological and immunohistochemical analysis of the osseous specimen revealed a soft-tissue tumor consistent with synovial sarcoma. The tumor was in direct contact with the neural elements. Resection with anything other than a marginal margin would have necessitated a resection of neural tissue, leading to a functional deficit. The oncology team and the family therefore planned a marginal resection with anticipated postoperative chemotherapy and radiation. A midline incision was made, ellipsing the previous biopsy incision. Sharp dissection was performed around the tumor, isolating the lesion with a cuff of 2 to 3 cm of paraspinal muscle tissue. This dissection was carried laterally to the lateral tip of the transverse processes of L2 through L5. Dissection was carried out on the anterior aspect of each transverse process to identify the lateral wall of the pedicles of L3 and L4, which were then osteotomized with a curved osteotome. The spinous processes and the laminae of L2 and L5 were resected. The tumor with its sheath of muscle was then elevated off the dura with use of a Penfield dissector to gently dissect dura from the tumor. Titanium clips were placed at the margins of the tumor resection to assist with postoperative radiation. Posterior reconstruction was accomplished with pedicle screws placed from L1 to S1, with autogenous iliac crest graft placed posterolaterally. Wound margins were approximated and closed without difficulty with a Hemovac drain (Zimmer, Warsaw, Indiana) in place. No muscle flap was needed to achieve closure. Because the tumor extended into the pedicle-body junction, an anterior corpectomy of L3 and L4 was performed immediately after the posterior procedure through a side-lying left retroperitoneal exposure. Reconstruction was accomplished with a humeral shaft allograft from L2 to L5 (Fig. 4). The estimated blood loss was 6000 mL for the entire procedure. Gross examination of the surgical specimen revealed a 10.0-cm lobulated soft-tissue mass weighing 310 g surrounded by a thin, fibrous pseudocapsule. Histologic sections consisted of a cellular proliferation of spindle cells arranged in fascicles with foci of myxoid change. The cells were associated with ectatic, thin-walled vascular channels with a prominent hemangiopericytomatous pattern. No epithelial component was identified. Brisk mitotic activity and foci of necrosis (comprising <5% of the total tumor volume) were noted, with a poorly differentiated morphology comprising <20% of the total tumor volume. The tumor extended to the lateral and deep margins and had infiltrated the surrounding connective tissue, including fascia and skeletal muscle, with foci of lymphovascular invasion (Figs. 3 and 5). The differential diagnosis of monophasic synovial sarcoma is broad, but the primary additional diagnostic considerations in this case included malignant peripheral nerve sheath tumor, malignant solitary fibrous tumor, leiomyosarcoma, and fibrosarcoma. Immunohistochemical analysis was performed on the resected specimen, and the tumor was negative for CD34 (My10; Becton-Dickinson, San Jose, California), providing no support for the diagnosis of solitary fibrous tumor. The tumor was negative for S100 (Dako, Carpinteria, California), muscle-specific actin (HHF35; Dako), and desmin (33; BioGenex, San Ramon, California), providing no support for neural, skeletal, or smooth muscle differentiation, respectively. The combination of membranous immunoreactivity for HBA71 (CD99) (013; Signet, Dedham, Massachusetts) and epithelial membrane antigen (E29; Dako) and cytoplasmic staining for CK7 (OV-TL; Dako) supported a diagnosis of synovial sarcoma. While peripheral primitive neuroectodermal tumor/Ewing sarcoma (pPNET/ES) also exhibits CD99 positivity, pPNET/ES lacks the prominent spindled morphology and hemangiopericytomatous vascular pattern seen in this case. Cytogenetic analysis confirmed the presence of a normal female karyotype. Despite the absence of the t(x;18)(p11.2;q11.2) gene mutation found in >90% of the cases of synovial sarcoma, the morphologic and immunohistochemical features best support the diagnosis of a poorly differentiated monophasic synovial sarcoma. On the second day of hospitalization, the patient had development of pulmonary edema, which responded well to diuretic therapy. The rest of the hospital stay was uneventful, and the patient was discharged home on the seventh day of hospitalization. At the time of the two-week follow-up, the patient was found to have made a full neurologic recovery, the incisions were healing well, and no pain medications were required. Radiation therapy and chemotherapy began one month after surgery with two cycles of Pediatric Oncology Group (POG) protocol #9553 chemotherapy. Each cycle consisted of 4.02 g of ifosfamide and 1 g of mesna on Days 1, 2, and 3. Forty milligrams of doxorubicin were administered on Days 1 and 2, and 2 mg of vincristine were given on Day 1. Two hundred milligrams of granulocyte-colony stimulating factor (G-CSF) were prescribed for seven days at the time of discharge. Following these initial cycles of chemotherapy, the patient underwent radiation therapy without chemotherapy with external beam radiation. A total dose of 5940 cGy was administered with progressively narrowed fields to reduce local nontarget tissue injury. After the completion of radiation treatment, the patient received an additional four cycles of chemotherapy. A routine screening laboratory examination demonstrated that chronic kidney disease had developed approximately five years postoperatively, believed to be a consequence of ifosfamide. Although the majority of the blood pressure readings remained normotensive, the patient was started on a low-dose angiotensin-converting enzyme inhibitor to control the blood pressure and to decelerate the progression of the renal failure. At the time of the most recent follow-up appointment, five years and nine months after surgery, the patient was able to achieve 80° of flexion and 20° of trunk extension and had 5/5 lower extremity strength bilaterally. She was without pain, had been participating in both soccer and ice skating, was working at a retail job, and was attending college classes. At the time of publication, the patient was receiving long-term monitoring in the Post-Treatment Sarcoma Clinic. This follow-up includes yearly physical examinations, regular monitoring of renal function, annual gadolinium-enhanced magnetic resonance imaging scans of the lumbar spine, and positron emission tomography-computed tomography, which to date have shown no evidence of local recurrence or distinct disease.
Proceed to Discussion >>Reference: Barus CE, Monsey RD, Kalof AN. Poorly differentiated synovial sarcoma of the lumbar spine in a fourteen-year-old girl. A case report. J Bone Joint Surg Am. 2009:91:1471-6. doi:10.2106/JBJS.H.00549
We know of only six reports on synovial sarcoma involving the lumbar spine in the English-language literature. Three of these reports involved pediatric patients, and only one report described the clinical course in any detail. This paucity of information is explained by the fact that synovial sarcoma at any location represents only 5.8% of all soft-tissue sarcomas in pediatric populations. The case described here represents the most comprehensive presentation of the clinical course, treatment, and outcome of a synovial sarcoma in the spine. Approximately 10% to 20% of synovial sarcomas invade adjacent bone. This was a prominent radiographic feature in the case of our patient, with destruction of the posterior facets and laminae as well as the L3 and L4 pedicles. The amount of osseous destruction and involvement as well as abutment against the thecal sac led to the decision to perform a marginal excision given the constraints of preserving neural tissue. The surgical technique allowed for gross total excision through the pseudocapsule. Histologically, there are three types of synovial sarcoma: biphasic, monophasic fibrous, and monophasic epithelial. An additional subtype of synovial sarcoma is the classification of "poorly differentiated," which may be superimposed on any of the other types as a histologic modifier. Because of the diversity of histologic features that may be present in association with a poorly differentiated synovial sarcoma, differentiating this entity from other small round-cell neoplasms may be difficult. Immunohistochemical and molecular genetic studies may be required to distinguish it from other possibilities such as malignant peripheral nerve sheath tumor, malignant solitary fibrous tumor, leiomyosarcoma, fibrosarcoma, peripheral primitive neuroectodermal tumor/Ewing sarcoma (pPNET/ES), and mesenchymal chondrosarcoma. Risk factors for poor outcomes such as metastasis or death include an age of more than twenty-five years, a tumor size of >5 cm, a tumor with >20% poor differentiation, and a primary tumor site in the lower extremities. The lungs (81.1%), lymph nodes (23%), and bone marrow (20%) are the most common sites of metastasis. The mean duration of disease from the onset of symptoms to death is approximately 6.5 years. The rate of survival in children is much better than that in adults, with a ten-year survival rate of 90% for those less than twenty years old as compared with only 25% for those more than forty years old. Synovial sarcoma typically reoccurs within two years, but it may do so as late as thirty-five years after treatment. Remission rates of 40% are possible when clear surgical margins are achieved or adjuvant radiation therapy is used. In the case of our patient, marginal resection and treatment with both chemotherapy and radiation therapy achieved more than five years of disease-free survival. Chemotherapy for the treatment of synovial sarcoma in general is somewhat controversial. Our patient had several risk factors for recurrence or metastasis, including the large size of the lesion, its poorly differentiated nature, and the treatment of the lesion with marginal resection. Given these risk factors, a sarcoma chemotherapy protocol was added to the postoperative treatment regimen. Our patient had an uncommon tumor presenting in a rare location. Our goal in presenting this case is to highlight for clinicians this anatomic site as a location for this lesion and to encourage its inclusion in the differential diagnosis for young patients with back pain and spinal masses. In addition, we provide an overview of the diagnostic considerations for this lesion and the decision-making process for the treatment of this lesion in this unique anatomic location.
Reference: Barus CE, Monsey RD, Kalof AN. Poorly differentiated synovial sarcoma of the lumbar spine in a fourteen-year-old girl. A case report. J Bone Joint Surg Am. 2009:91:1471-6. doi:10.2106/JBJS.H.00549

Synovial sarcoma
Rhabdomyosarcoma
Malignant nerve sheath tumor
Leiomyosarcoma
Fibrosarcoma