A Seventeen-Year-Old Boy with a Ten-Month History of Swelling of the Right Foot
September 7, 2011
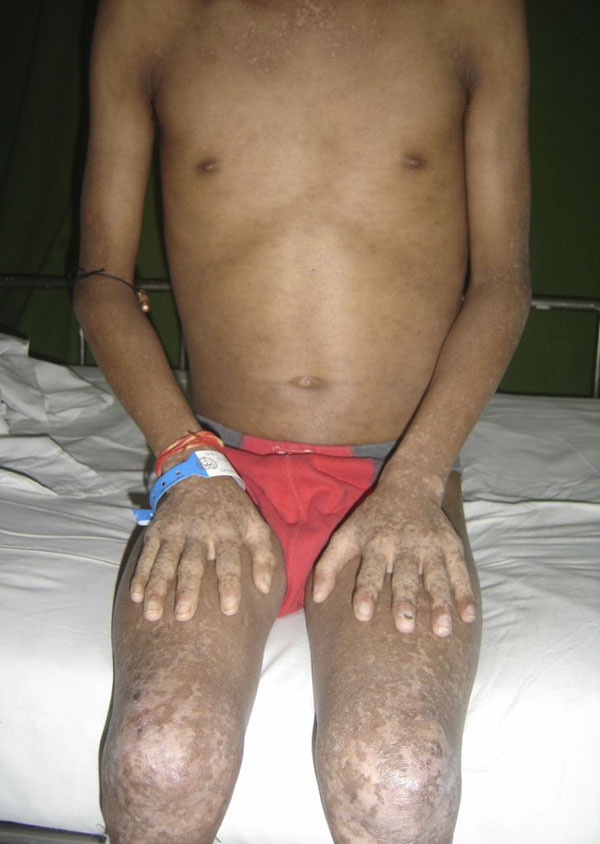
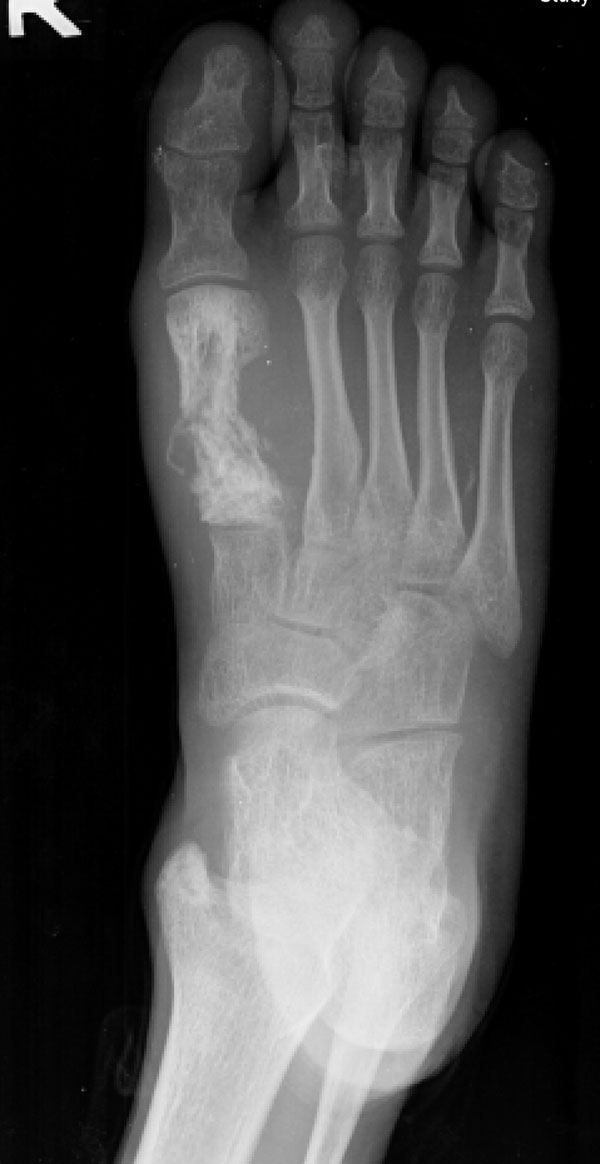
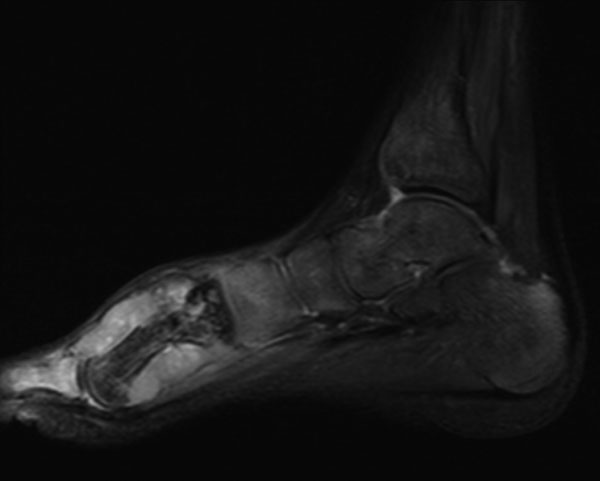
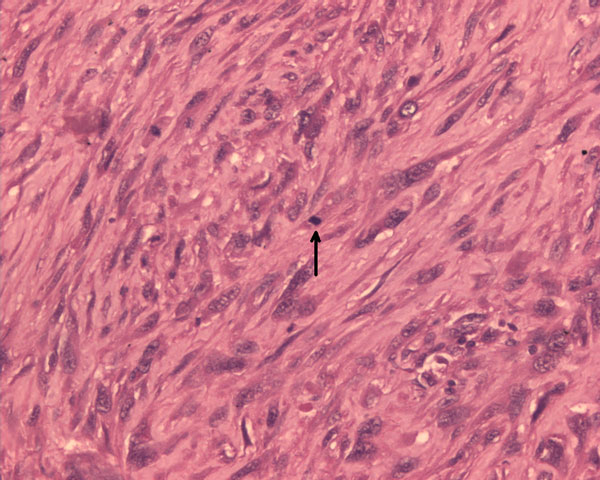
 Fig. 1
Fig. 1 Fig. 2
Fig. 2 Fig. 3
Fig. 3 Fig. 4
Fig. 4 Fig. 4
Fig. 4 Fig. 5
Fig. 5A seventeen-year-old boy presented with a ten-month history of progressive swelling of and pain in the first ray of the right foot. He was the fourth child born to consanguineous parents. At the age of six months, he had development of facial erythema, which spread to the arms, legs, and buttocks, later evolving into a reddish-brown rash with hypopigmented lesions. The skin lesions were less prominent on the trunk. He had delayed developmental milestones and was a slow learner. There was no family history of cancer, dermatological problems, or learning disabilities. Examination revealed a height of 145 cm and a weight of 30 kg (both below the third percentile for age). Cutaneous examination showed reticulated, pigmented macules with atrophy and telangiectases predominantly on the cheeks, elbows, knees, thighs, buttocks, and dorsal parts of the hands and feet (Fig. 1). The patient had punctate keratoderma of the hands and feet and hyperkeratosis of the palms and soles. The left fourth fingernail was rudimentary, and the patient had sparse scalp hair, eyebrows, and eyelashes. His mental and verbal abilities were those of a nine-year-old child. Examination of the eyes and teeth revealed normal findings. On the basis of the characteristic appearance and evolution of the skin lesions, a diagnosis of Rothmund-Thomson syndrome was made. The right foot showed a fusiform swelling arising from the distal metaphysis of the first metatarsal and extending into the first web space. The swelling was mildly tender; was palpable on the medial, plantar, and dorsal aspects of the foot; and had a varied consistency. A radiograph of the right foot showed marked mottled destruction of the first metatarsal, characterized by sclerosis and interspersed irregular lytic areas, soft-tissue swelling, and osteopenia of the other bones of the foot (Fig. 2). The chest radiograph revealed normal findings. Magnetic resonance imaging revealed soft-tissue extension of the tumor into the adjacent muscles (the abductor hallucis and flexor hallucis longus) and into the first web space, extending inferiorly into the subcutaneous layer of the sole but not into the adjacent joints (Fig. 3). A negative bone scan ruled out any other sites of osseous involvement. Chromosomal analysis of twenty leukocyte metaphases from the peripheral blood revealed a normal male karyotype (46, XY). After informed consent was obtained, the samples from the patient and his parents were sent to a laboratory conducting research on Rothmund-Thomson syndrome. Sequencing of the RECQL4 gene, performed by the Medical Genetics Laboratory at Baylor College of Medicine (Houston, Texas), revealed that the patient was homozygous for a g.4428_5437 deletion mutation. The parents were found to have one copy each of the same mutation of the RECQL4 gene. These findings confirmed the diagnosis of Rothmund-Thomson syndrome. Treatment of the foot was delayed as the patient had development of fever with chills and had splenomegaly on abdominal examination. Examination of the blood showed pancytopenia, a total white blood-cell count of 1 × 109/L, a platelet count of 60 × 109/L, and a packed cell volume of 24%. Bone marrow trephine biopsy showed mildly hypercellular marrow with erythroid hyperplasia. The pancytopenia was eventually attributed to hypersplenism due to infectious mononucleosis, which was confirmed by a cervical lymph node biopsy showing paracortical expansion and marked immunoblastic proliferation. Once these medical issues had been resolved, a needle biopsy of the first metatarsal was done (Fig. 4).
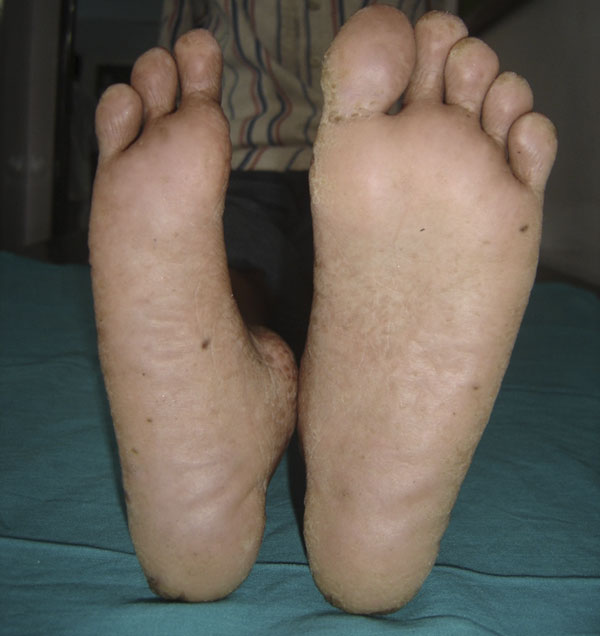
Needle biopsy of the tumor confirmed osteosarcoma. The patient underwent amputation of the first and second rays, with the removal of the medial and middle cuneiforms. On histopathological examination of the resected tissue, the tumor was classified as high grade because of high cellularity, nuclear pleomorphism, and mitotic activity (Fig. 4). Osteoid production by tumor cells was seen focally. Reconstruction at the midfoot included transfer of the tibialis anterior and extensor hallucis longus to the dorsum of the navicular. On the plantar aspect of the foot, the stump of the flexor hallucis longus tendon was attached to the navicular, the flexor digitorum tendon was attached to the remnant of the transverse plantar ligament near the lateral cuneiform, and the peroneus longus tendon was attached to the base of the third metatarsal. The margins of the specimen were free of tumor. After ray amputation, in view of persisting hypersplenism, a splenectomy was performed and the blood cell counts subsequently returned to normal. Postoperative chemotherapy consisting of six cycles of cisplatin and doxorubicin was planned; however, only three cycles could be delivered because of recurrent episodes of febrile neutropenia. Each cycle of chemotherapy was accompanied by injections of granulocyte colony-stimulating factor. For the third cycle, the doses of the cisplatin and doxorubicin were reduced by 25%. At the end of the third cycle, because of persistent neutropenia, chemotherapy was discontinued. After surgery, the limb was immobilized in a below-the-knee cast following suture removal for a period of six weeks. At six weeks, the patient underwent physiotherapy, and a shoe filler prosthesis was provided. The patient was satisfied with the functional outcome, and he was able to walk without support after six weeks. At twenty-six months, he was doing well, with no evidence of recurrent disease. The appearance of his foot at that time is shown in Figure 5.
Proceed to Discussion >>Reference: Padhy D, Madhuri V, Pulimood SA, Danda S, Walter NM, Wang LL. Metatarsal osteosarcoma in Rothmund-Thomson syndrome. A case report. J Bone Joint Surg Am. 2010;92:726-30.
Rothmund-Thomson syndrome is a rare autosomal recessive atrophic dermatosis with an onset in infancy. The chronic pattern of reticulated hypopigmentation and hyperpigmentation, punctate atrophy, and telangiectases, collectively known as poikiloderma, persists throughout life. Rothmund-Thomson syndrome is associated with abnormalities of the RECQL4 gene. Entire gene sequencing detects mutations in approximately 66% of affected individuals. Genotype-phenotype analysis has shown clearly that the development of osteosarcoma is associated with the presence of mutations that are predicted to result in loss of RECQL4 protein function. Most of the reported deleterious RECQL4 mutations are nonsense, splicing, or frameshift mutations. The mutation in our patient, a deletion spanning exon 14 to exon 18 of the RECQL4 gene, has not been previously reported to our knowledge and is unique because of its large size (1010 base pair deletion). Generalized skeletal dysplasia, including abnormal trabeculation, malformed and fused bones, radial ray defect, and delayed bone formation, has been described in association with Rothmund-Thomson syndrome. The first case of osteosarcoma associated with Rothmund-Thomson syndrome was reported in a thirteen-year-old boy in 1980. Wang et al., in a cohort of forty-one patients with Rothmund-Thomson syndrome, found the prevalence of osteosarcoma to be 32%. The average age of patients who have development of osteosarcoma in association with Rothmund-Thomson syndrome is younger than that of patients who have osteosarcoma in the general population, and the incidence is equal among boys and girls. Sites of osteosarcoma in patients with Rothmund-Thomson syndrome have been reported to be similar to those in the general population: the metaphyseal regions of long bones, most commonly the distal part of the femur and the proximal part of the tibia. The treatment response and overall outcomes in patients with Rothmund-Thomson syndrome with osteosarcoma appear to be similar to those in the general population. Therefore, every attempt should be made to deliver all chemotherapy courses on time in order to optimize survival outcome for these patients. In our patient, this was not feasible because of prolonged fever and pancytopenia, presumably due to infectious mononucleosis, which required splenectomy. The pancytopenia resolved, but the additional surgery led to further treatment delays. To our knowledge, infectious mononucleosis and splenomegaly have not previously been associated with Rothmund-Thomson syndrome. In terms of local control in our patient, radical resection by means of ray amputation was possible because of the metatarsal location of the tumor. Amputation around the midtarsal region usually results in postoperative deformities due to muscle imbalance. Extended amputation of the first ray results in cutting of slips of the tendons of the tibialis posterior from the medial cuneiform, the tibialis anterior and the peroneus longus from the first metatarsal, and the extensor and flexor hallucis longus from the distal phalanx of the hallux. Second ray amputation causes an additional loss of the first slips of the tendons of the flexor and extensor digitorum. We anticipated that this type of muscle imbalance, combined with unopposed triceps surae activity, would result in equinovalgus deformity with loss of the longitudinal arch of the foot. Proximal reattachment of the tendons in a balanced fashion as was done in this case is similar to reattachment of the tibialis anterior tendon as described for a midtarsal amputation. To our knowledge, such a reconstruction following the extended amputation of the first two rays as was done in our patient has not been described in the English-language literature. This restoration of muscle balance may have been responsible for the favorable cosmesis and function of the foot postoperatively. A prolonged period of follow-up after the treatment of osteosarcoma in patients with Rothmund-Thomson syndrome is recommended in order to look not only for metastasis but also for the occurrence of a second cancer, which has been reported in the literature. Metastasis is predominantly pulmonary. Patients should be screened periodically with use of chest radiographs, computed tomography, or bone scanning as required for any other osteosarcoma, and long-term follow-up for future skeletal, hematological, and dermatological malignant lesions should be carried out. In summary, we present what we believe to be the first case of Rothmund-Thomson syndrome with a novel homozygous deletion in the RECQL4 gene, the largest deletion reported to date for this condition. Other highlights of the case are the unusual site of osteosarcoma in the first metatarsal, the ray amputation with tendon transfers to balance the muscle pull on the foot, and complexities in the treatment of the tumor because of the incidental occurrence of infectious mononucleosis and hypersplenism.
Reference: Padhy D, Madhuri V, Pulimood SA, Danda S, Walter NM, Wang LL. Metatarsal osteosarcoma in Rothmund-Thomson syndrome. A case report. J Bone Joint Surg Am. 2010;92:726-30.

Amputation of the first and second rays with clear margins
Syme amputation
Aggressive bone and soft tissue debridement with planned long-term antibiotics and likely repeat debridement
Systemic corticosteroid treatment with serial imaging studies to evaluate steroid effect