A Twenty-nine-Year-Old Woman with Right Knee Pain for Over One Year
August 3, 2011
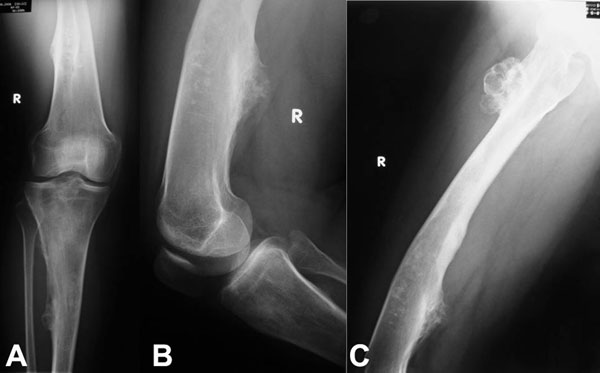
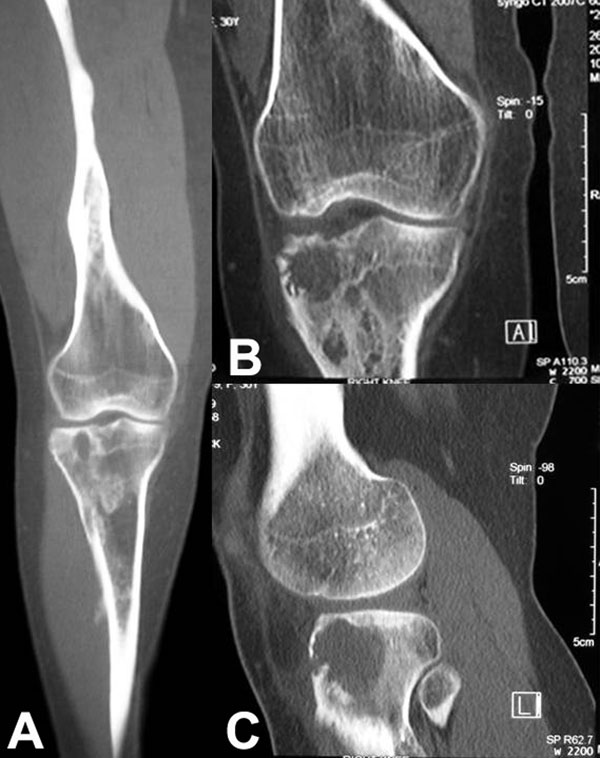
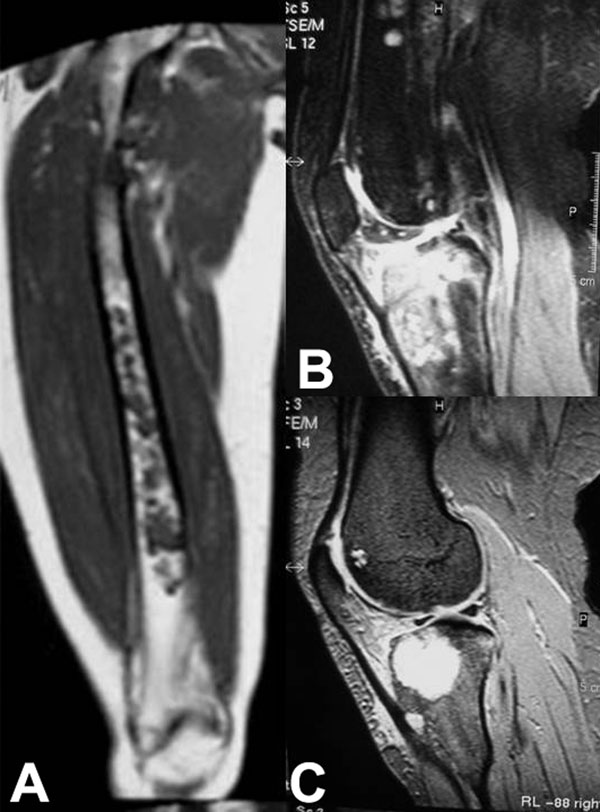


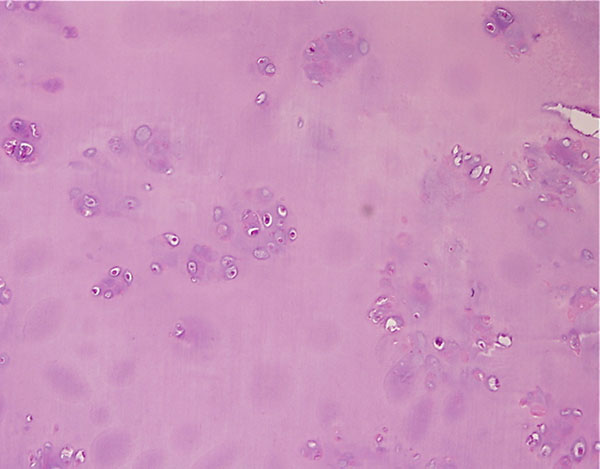
 Fig. 1
Fig. 1 Fig. 2
Fig. 2 Fig. 3
Fig. 3 Fig. 4-A
Fig. 4-A Fig. 4-B
Fig. 4-B Fig. 4-C
Fig. 4-C Fig. 4-A
Fig. 4-A Fig. 4-B
Fig. 4-B Fig. 4-C
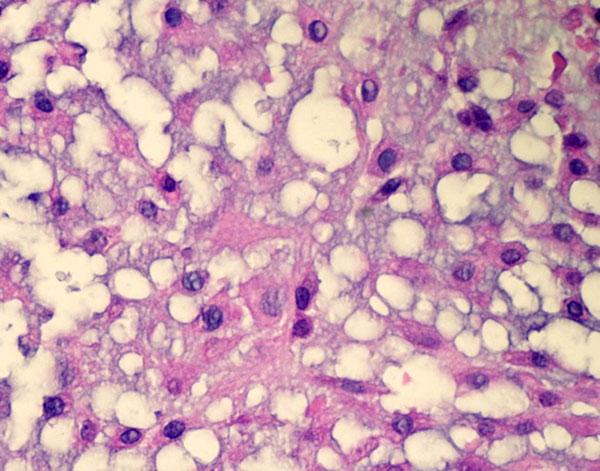
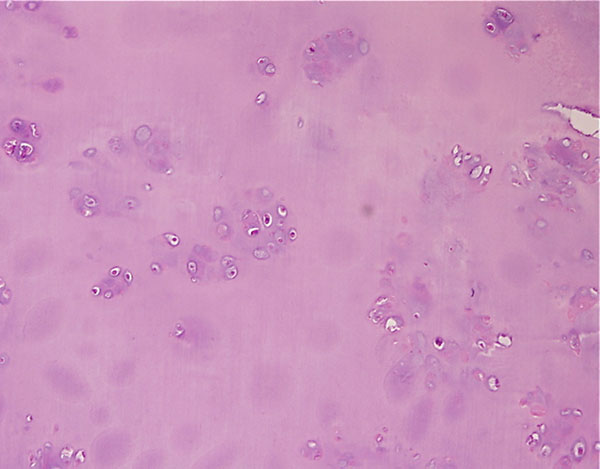
Fig. 4-CA twenty-nine-year-old woman was admitted to our institution complaining of pain localized to the right knee for over one year. Physical examination showed localized tenderness at the anterolateral aspect of the proximal part of the right tibia. There was no palpable mass. Anteroposterior and lateral radiographs of the right knee showed skeletal osteochondromas and enchondromas of the right femur and tibia as well as an osteolytic lesion involving the lateral tibial condyle (Fig. 1). A computed tomography scan showed a cystic osteolytic lesion involving the right proximal tibial metaphysis with erosion of the anterior and lateral cortex, and an adjacent proximal tibial enchondroma (Fig. 2). Magnetic resonance imaging revealed a destructive lesion of the lateral aspect of the right proximal tibial metaphysis, with erosion of the anterior and lateral cortex, without an associated soft-tissue mass (Fig. 3). A bone scan revealed increased radioisotope uptake in the proximal part of the right tibia and at the region of the proximal aspect of the right femur, probably in the lesser trochanter. A computed tomography scan of the chest was negative. Routine laboratory data were within normal limits. Although radiographs of the pelvis and hands were normal, on the basis of the synchronous occurrence of skeletal osteochondromas and enchondromas, the diagnosis of metachondromatosis was established. A computed tomography-guided trephine biopsy of the lesion was performed with a later open biopsy. Cultures of the specimen were negative. Histological findings are shown in Figures 4-A, 4-B, and 4-C.
The initial trephine biopsy demonstrated some features suggestive of chondrosarcoma, but a definitive diagnosis of malignancy could not be made. Accordingly, through an anterolateral approach, complete curettage of the lesion, with use of a high-speed burr, and application of phenol and acrylic bone cement as a bone-void filler were accomplished. Gross examination of the curetted specimen showed translucent, whitish, vaguely nodular tissue fragments and blood clots. Final histological examination showed a cartilaginous neoplasm consistent with an intermediate-grade (grade-2) chondrosarcoma (Fig. 4-B); focally, hypocellular areas with a lobular architecture, mild cytologic atypia, and chondroid matrix reminiscent of enchondroma were also observed (Fig. 4-C). A noncartilaginous component or abrupt transition between the two tissue types suggestive of dedifferentiation was not observed. If the diagnosis of grade-2 chondrosarcoma had been established preoperatively, wide local resection with reconstruction would have been recommended. This patient was offered further surgical treatment when the histological diagnosis was established, but she opted for careful follow-up at this time. Seven months postoperatively, the patient was well with no evidence of local recurrence or metastasis.
Proceed to Discussion >>Reference: Mavrogenis AF, Skarpidi E, Papakonstantinou O, Papagelopoulos PJ. Chondrosarcoma in Metachondromatosis: A Case Report. J Bone Joint Surg Am. 2010; 92:1507-1513.
Primary chondrosarcomas arise de novo, whereas secondary chondrosarcomas originate in a preexisting lesion such as an enchondroma or osteochondroma. Malignant transformation of an enchondroma to chondrosarcoma is rare. The risk of chondrosarcoma is greater in patients with enchondromatosis syndromes, such as Ollier disease and Maffucci syndrome, and in those with hereditary multiple exostoses. Therefore, the association of malignant degeneration with still another dysplastic cartilaginous disorder such as metachondromatosis is not totally unexpected. To our knowledge, however, this is the first documented case of the malignant transformation of a metachondromatosis-associated enchondroma to chondrosarcoma. Metachondromatosis is easily differentiated from other conditions that feature multiple chondromatous lesions, including hereditary multiple exostoses and enchondromatoses. Hereditary multiple exostoses is an autosomal dominant disorder in which osteocartilaginous exostoses are present. Hereditary multiple exostoses may have considerably bizarre appearances and often produce shortening and deformity of the long bones (particularly the distal end of the radius and ulna). This condition manifests during the first or second decade of life as palpable osseous protuberances with secondary deformity. Additionally, malignant transformation of these exostoses has been described. Molecular analysis has implicated loss of tumor suppression factors in the pathogenesis. Enchondromatosis, or multiple enchondromas, occurs in three different conditions, including metachondromatosis; multiple enchondromatoses, or Ollier disease; and Maffucci syndrome. The most common entity is Ollier disease, a nonhereditary failure of cartilage ossification, involving typically unilateral enchondromata without exostoses. It usually becomes evident before puberty and is frequently unilateral, leading to shortening of the limbs. This entity, however, shares with metachondromatosis the characteristic of frequent spontaneous resolution of osseous lesions. In Ollier disease, the prevalence of malignant transformation has been reported to range from 30% to 50%. Maffucci syndrome is also a nonhereditary syndrome that is rarer than Ollier disease. It is characterized by multiple enchondromas as well as multiple soft-tissue cavernous hemangiomas, and less commonly, lymphangiomas. There is also a higher risk of malignant transformation of Maffucci syndrome-associated enchondromas to sarcomas compared with those of Ollier disease or solitary enchondromas. The metachondromatosis-associated enchondromatous lesions, in contrast to enchondromas in patients with Ollier disease and Maffucci syndrome, mainly affect the iliac crests and the metaphyses of the long bones. Both Maffucci syndrome and Ollier disease are also associated with an increased prevalence of juvenile granulosa cell tumor of the ovary. Patients with Maffucci syndrome also have an increased prevalence of malignant lesions other than musculoskeletal malignant tumors, including gliomas, gastrointestinal adenocarcinomas, pancreatic carcinomas, and ovarian tumors. Genochondromatosis and dysspondyloenchondromatosis are familial conditions with enchondromas but lack exostoses and periarticular ossified lesions. Three individuals of a family with metachondromatosis and clinical features resembling those of the trichorhinophalangeal syndrome have been reported. In the patient in the current report, although radiography of the pelvis and hands did not show enchondromas or skeletal osteochondromas, the synchronous occurrence of such lesions suggested the diagnosis of metachondromatosis since the diagnosis of any of the above dysplastic chondromatous conditions could not be established. Histologically, skeletal osteochondromas and metachondromatosis display the same characteristic growth plate architecture. However, at the molecular level, metachondromatosis is a separate entity. By using cDNA microarray analysis, Bovée et al. found that metachondromatosis clusters separately from skeletal osteochondromas and growth plates. Although not performed in our patient, genetic testing is useful to establish Exostosin (EXT) gene abnormality and mutations in patients with multiple or solitary osteochondromas, enchondromatosis, and metachondromatosis. To date, two Exostosin genes (EXT-1 and EXT-2) are known to be involved in the formation of osteochondromas, and forty-nine different EXT-1 and twenty-five different EXT-2 mutations have been found in patients with hereditary multiple osteochondromatosis; there is evidence that mutations in these two genes are responsible for >70% of the hereditary multiple osteochondromatosis cases. The EXT-1 and EXT-2 genes involved in hereditary multiple osteochondromatosis, and downregulated in solitary osteochondromas, are normally expressed in metachondromatosis. The EXT genes are involved in heparan sulfate biosynthesis, important for Indian hedgehog/parathyroid hormone-like hormone (Ihh/PTHLH) growth plate signaling pathways. The Ihh/PTHLH signaling molecules are expressed in metachondromatosis as shown by both quantitative polymerase chain reaction and immunohistochemistry, suggesting that this pathway is active. This is in contrast to osteochondromas, in which PTHLH signaling is downregulated. Besides EXT-1 and EXT-2 genes, three other genes (EXT-like genes) showing homology to the EXT genes have been identified: the EXTL-1 on chromosome 1p36, the EXTL-2 on chromosome 1p11-p12, and the EXTL-3 on chromosome 8p12-p22. To date, only the function of EXTL-2 has been determined and, not surprisingly, this gene was also shown to be a transferase involved in heparan sulfate biosynthesis. Although the EXT and the EXTL-2 genes act in the same biological pathway, involvement of the EXT-like genes in the formation of osteochondromas has not yet been proven. It may very well be that mutations in EXTL-2 have a much more severe result, as this gene acts upstream of the EXT genes in heparan sulfate biosynthesis. Furthermore, at present none of the other EXT-like genes have been proven to be involved in the development of osteochondromas, and their possible involvement in any disorder remains to be elucidated. The natural history and the risk for malignant degeneration of any single metachondromatosis lesion cannot be predicted. Since spontaneous regression of some osteochondromas may be expected, not all asymptomatic lesions should be treated surgically. Close monitoring is necessary in patients with large, growing long-bone osteochondromas (nerve and vascular compression) and in patients with femoral head involvement. In the case of the current patient, we acknowledge that the treatment of an intermediate-grade chondrosarcoma with intralesional surgery is inappropriate; because of the initial histological diagnosis of a low-grade tumor and the young age of the patient, marginal excision, chemical ablation with phenol, and cementation were done. After definitive diagnosis of a grade-2 lesion, the patient was informed regarding immediate reoperation and agreed to undergo close follow-up evaluation with use of magnetic resonance imaging; in the case of local recurrence, wide surgical excision will be performed. However, the purpose of this study was not to present the treatment and prognosis of primary or secondary chondrosarcomas but to present the malignant transformation of a metachondromatosis-associated enchondroma to chondrosarcoma.
Reference: Mavrogenis AF, Skarpidi E, Papakonstantinou O, Papagelopoulos PJ. Chondrosarcoma in Metachondromatosis: A Case Report. J Bone Joint Surg Am. 2010; 92:1507-1513.

Wide local resection with allograft or tumor prosthesis reconstruction
Curettage of the lesion with iliac crest bone graft
Complete curettage of the lesion, phenol application, and cementation
Knee disarticulation amputation