An 8-Month-Old Boy with Tenderness and Weakness in the Upper Arm
June 20, 2018
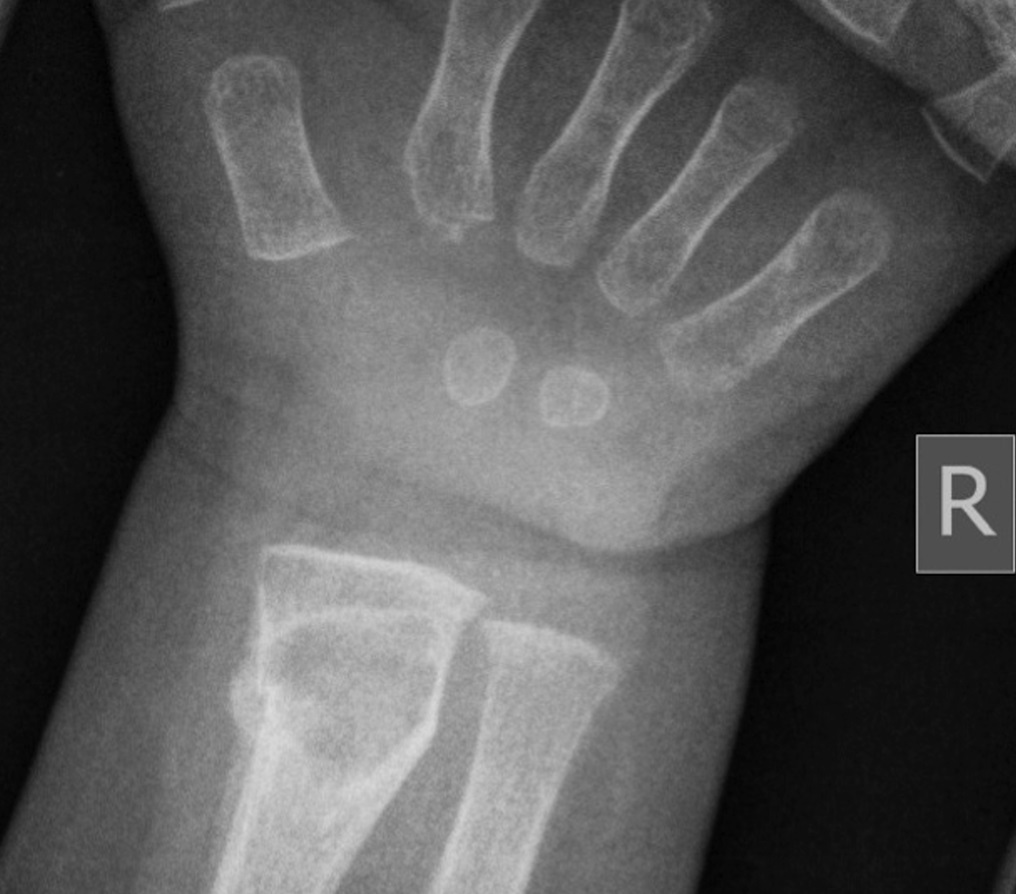
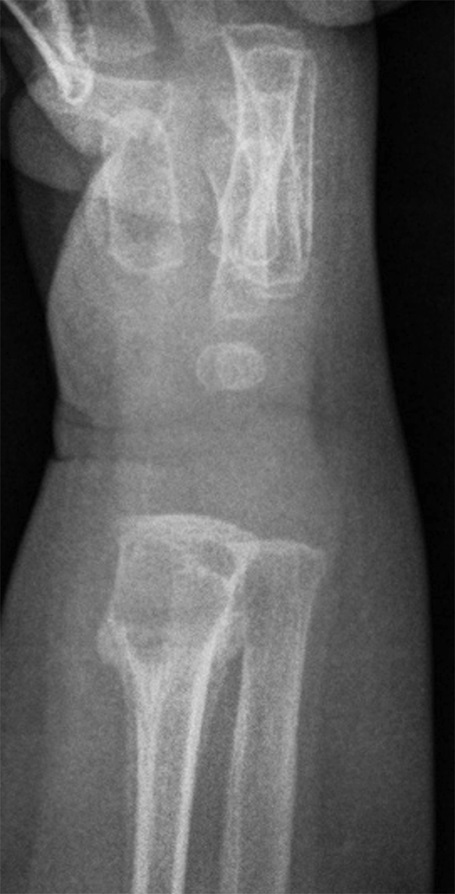
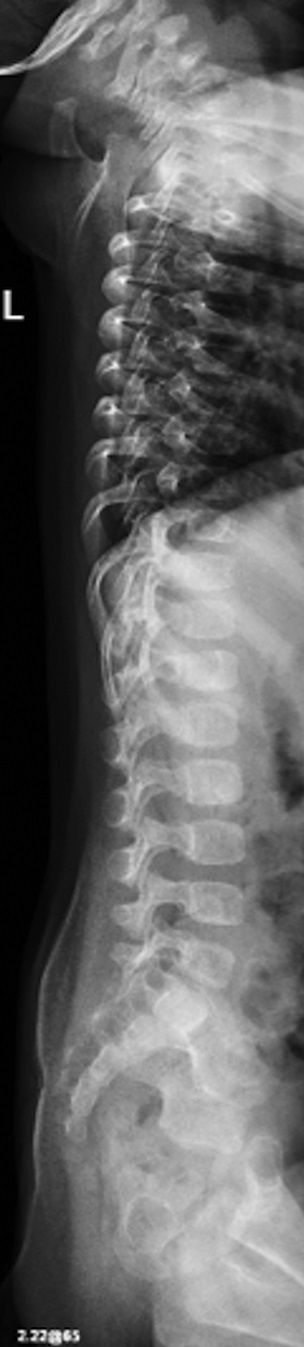
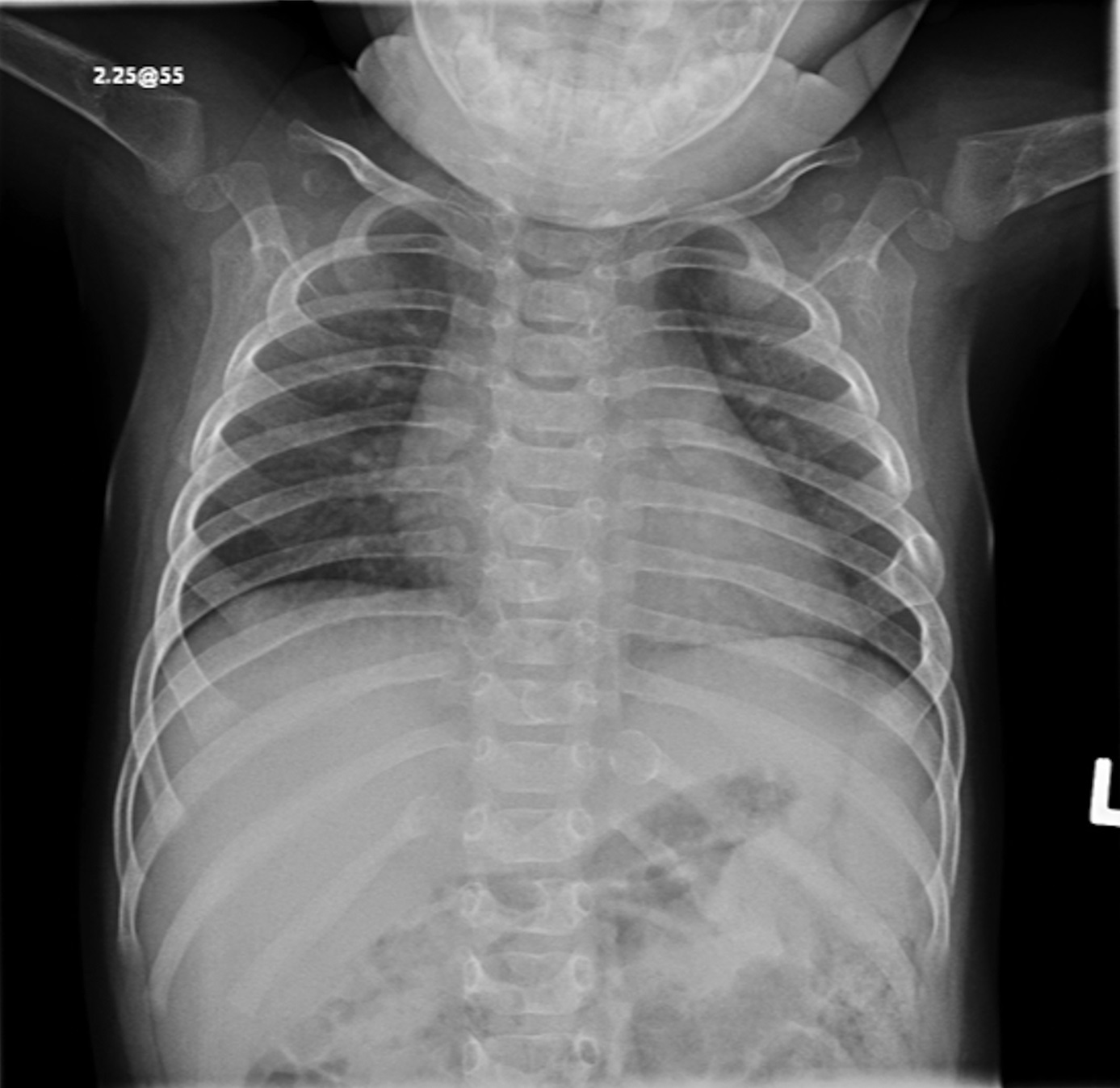
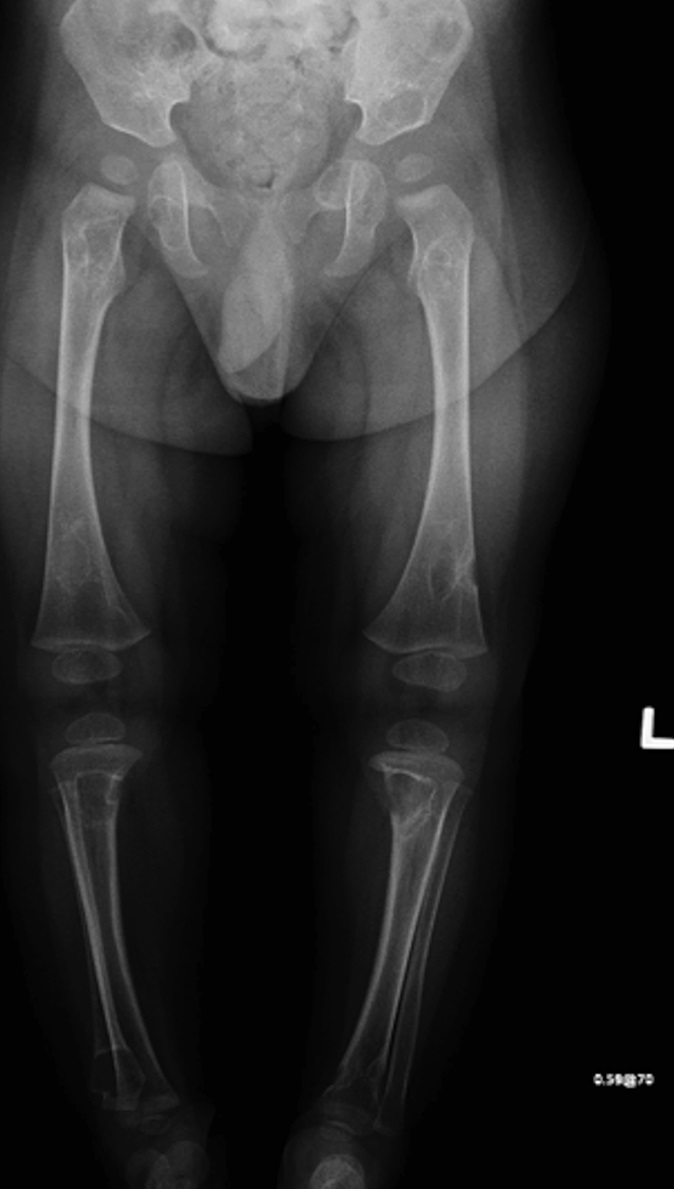
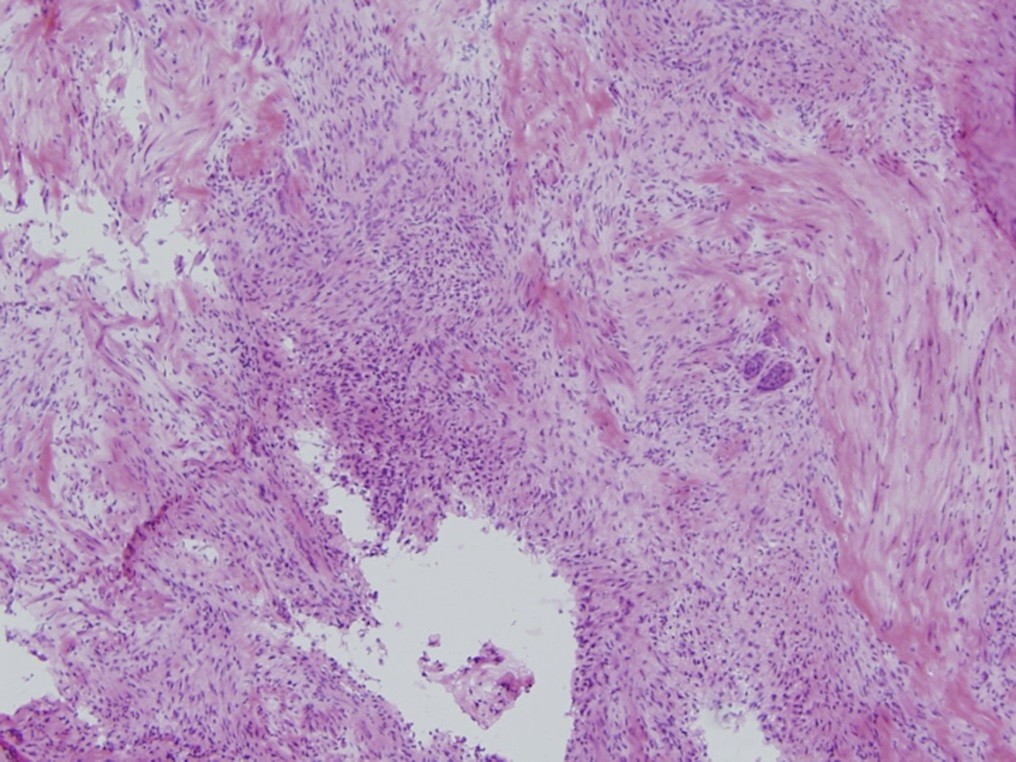


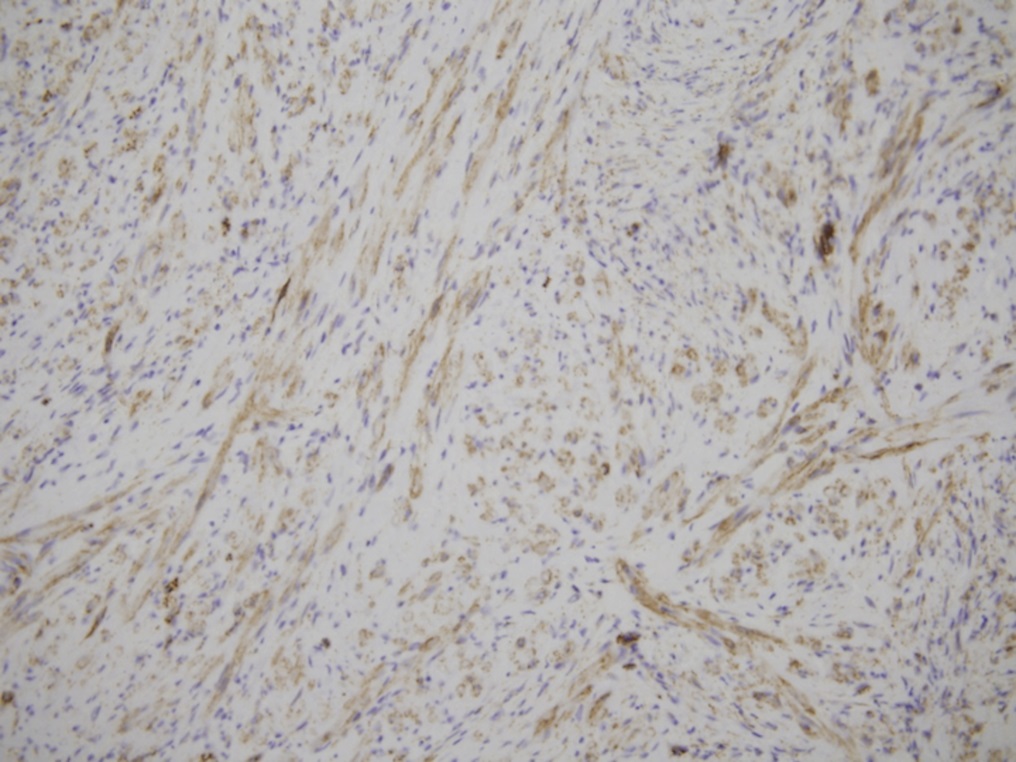
 Fig. 1-A
Fig. 1-A Fig. 1-B
Fig. 1-B Fig. 2-A
Fig. 2-A Fig. 2-B
Fig. 2-B Fig. 2-C
Fig. 2-C Fig. 3-A
Fig. 3-A Fig. 3-B
Fig. 3-B Fig. 3-C
Fig. 3-C Fig. 3-D
Fig. 3-D Fig. 4-A
Fig. 4-A Fig. 4-B
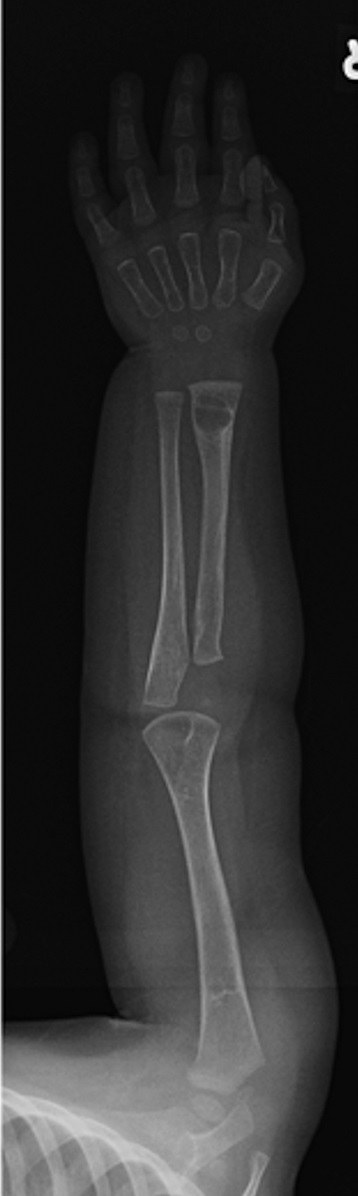
Fig. 4-BAn otherwise healthy 8-month-old boy presented to the children’s emergency department with decreased use of his right upper extremity. Physical examination revealed tenderness of the distal aspect of the right radius. Radiographs revealed a large lytic lesion of the metaphysis with well-defined margins associated with a nondisplaced pathologic fracture (Fig. 1). The patient was treated with immobilization in a below-the-elbow cast. Given the age of the patient, the most likely differential diagnosis of a lytic bone lesion included fibromatosis, metastatic neuroblastoma, and Langerhans cell histiocytosis. A skeletal survey showed lytic lesions with well-defined sclerotic margins in the proximal aspects of both humeri; distal aspects of both radii; left ilium; left superior pubic ramus; right ischium; proximal and distal aspects of both femora; proximal and distal aspects of both tibiae; distal aspect of the right fibula; 5th, 10th, and 11th ribs; and T7, T8, T9, and T12 vertebrae (Fig. 2). The results of an abdominal ultrasound examination were normal. The results of a blood analysis, including white blood-cell count, hemoglobin, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), creatinine, and liver and thyroid function tests, were all within normal limits. A detailed physical examination revealed no lymphadenopathy or other concerning features. A well-defined, mobile soft-tissue mass was found in the left axilla and was believed to be a lipoma.
Biopsies of 2 of the lesions in the left tibia revealed that one was a lytic lesion typical of the majority found in the patient, and the other appeared to have more sclerosis at one of the margins. The axillary soft-tissue mass was excised.
A frozen section of the bone biopsy specimens demonstrated fascicles of spindle cells in a collagenized background, with no evidence of atypia or malignancy (Fig. 3-A). On permanent sections, the 2 sampled lesions showed similar features, characterized by uniform, monotonous, fascicular proliferation of spindle-shaped cells arranged focally in a distinct nodular pattern (Fig. 3-B). The spindle cells showed a fibrillary cytoplasm typical of myofibroblasts and contained tapered, bland nuclei. A collagenous and focally myxoid extracellular matrix was also present. No slit-like (“stag-horn”) hemangiopericytic-pattern blood vessels were seen, and there was no evidence of atypical mitosis, necrosis, cellular pleomorphism, increase in eosinophils, or malignant “osteoid” formation. Fragments of otherwise unremarkable bone trabeculae were also present. Immunohistochemistry showed lesional cells positive for actin and negative for desmin (Fig. 3-C), CD34, and the myogenic markers myoD1 and myogenin. This staining pattern is typical of cells showing myofibroblastic differentiation. No cells that were positive for both S100 and CD1a, as might be seen in Langerhans cell histiocytosis, were identified. The Ki67 proliferation marker was low (1% to 2%). Beta-catenin was expressed variably at a low percentage (up to 15%) in the nuclei (Fig. 3-D), and there was ubiquitous cytoplasmic staining. The soft-tissue mass from the axilla had macroscopic and microscopic features in keeping with a lipoma.
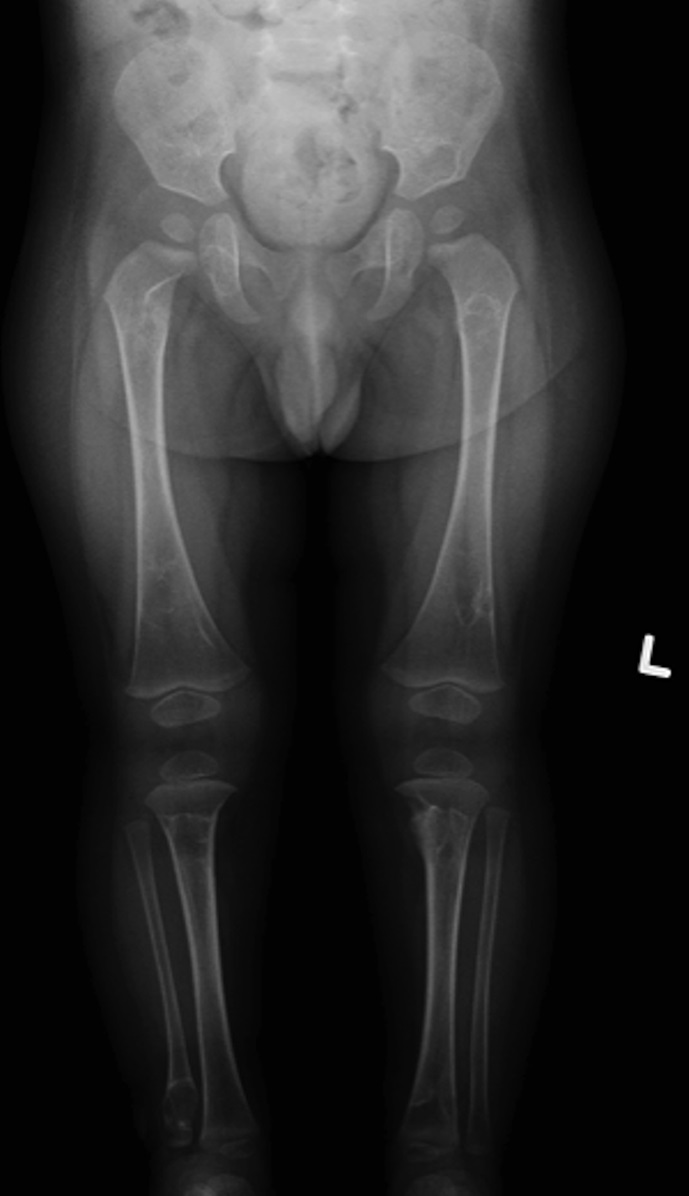
The patient was diagnosed with infantile myofibromatosis (IM). A computed tomography (CT) scan of the chest, performed to complete the systemic work-up for visceral involvement, revealed no abnormalities. A repeat skeletal survey performed approximately 8 weeks after presentation demonstrated lesions in various stages of regression, with some lesions decreasing dramatically in size and others maintaining their dimensions. No new lesions were observed (Figs. 4-A and 4-B). A third skeletal survey performed approximately 7 months after presentation showed further regression overall and no evidence of new lesions. At 1 year after presentation, the patient continued to reach appropriate milestones with no signs of pain, irritation, or deformity in association with the osseous lesions.
Proceed to Discussion >>Reference: McCammon J, Stefanovici C, Martin RK, Larouche P. Multiple bone lesions in an 8-month-old child presenting with pathologic fracture: a rare case of solely osseous multicentric infantile myofibromatosis. JBJS Case Connect. 2016 Apr-Jun;6(2):e42.
IM is a rare and benign myofibroblastic lesion that may occur in either a solitary or multicentric form. The incidence of IM is unknown, but the solitary form is believed to be among the most common fibrous diseases of infancy and early childhood.
Although rarely described, pathologic fracture may be the presenting feature in a child with myofibromatosis. Brill et al. described a patient with multicentric IM who presented with bilateral pathologic femoral fractures. Wu et al. recently reported the case of a 9-year-old girl with a pathologic diaphyseal humeral fracture.
The radiographic differential diagnosis of multiple osseous lesions in an infant includes Langerhans cell histiocytosis, infection, metastatic neuroblastoma, and fibrous tumors. The pathologic differential diagnosis includes IM, desmoplastic fibromas, low-grade fibrosarcomas, fibrous dysplasia, and non-ossifying fibromas.
IM lesions are typically discovered within the first 2 years of life, often in neonates. In a recent series of 28 infants and children, the mean age at onset was 3.1 years for the solitary form and 27 days for the multicentric type. A review by Levine et al. revealed 30 cases of the multicentric type with a median age at onset of 2 months and >100 solitary lesions with a median age at onset of 3 months.
Both autosomal-dominant inheritance with variable penetrance and autosomal-recessive inheritance have been described.
Radiographs of IM osseous lesions show a lucent region with marginal sclerosis. The lesions are typically located in the metaphysis, with sparing of the region immediately adjacent to the physis. CT characteristics include a mass with peripheral enhancement and calcifications. Magnetic resonance imaging (MRI) reveals low signal intensity on T1 weighting, with high or low signal on T2 weighting. Histology demonstrates fascicles of myofibroblastic, bland spindle-shaped cells in a whorled collagenized background, with a pericytic vascular pattern often present. Immunohistochemistry is positive for smooth muscle and muscle-specific actins, but negative for S100, desmin, and CD34. Beta-catenin has variable expression in the osseous lesions, with nuclear expression in the soft-tissue counterpart of up to 80% to 90%.
The solitary form of congenital fibromatosis occurs more commonly than the multicentric form, with ranges from 50% to 80% having been described. The solitary form typically presents with a single cutaneous nodule. These nodules have been described as firm and flesh-colored or purple and can be superficial or deep in the soft tissues. It is uncommon for lesions to be found in bone alone, with the rate reported to be around 5%, and the majority occur in craniofacial bones.
In multicentric IM, lesions typically affect the cutaneous and/or subcutaneous tissues, muscles, and bones. Published series indicate that 17% to 57% of patients with multicentric IM who do not have visceral involvement have lesions involving bone.
In 1982, Brill et al. summarized 39 cases of congenital generalized fibromatosis (CGF), which is believed to be the same entity as IM. Of the 39 cases, 24 had osseous involvement, but there were no recorded cases of CGF with only osseous involvement. In a more recent review, Levine et al. summarized 30 cases of multicentric IM with or without visceral involvement. Review of these cases revealed only 1 in which involvement was only osseous; all other cases had lesions that involved skin and soft tissue or organs.
Visceral involvement has been reported to occur in 27% to 73% of patients with multicentric IM. Gopal et al. reported 76% mortality in patients with visceral involvement. The seriousness of the possibility of visceral involvement demands the search for evidence of systemic disease in general and visceral involvement in particular, including chest radiography, abdominal ultrasonography, chest and abdominal CT, echocardiogram, a skeletal survey, and biopsies.
When involvement is nonvisceral, treatment may consist of careful observation, as most lesions spontaneously regress, but the exact time frame of regression is not described in the literature. It has been proposed that this spontaneous regression is mediated by massive apoptotic cell death. There have been case reports of solitary lesions treated with curettage and bone-grafting. This resulted in recurrence with eventual healing in 1 case and more immediate healing in 2 other cases. To our knowledge, no cases involving this treatment in patients with multiple osseous lesions have been described. In cases of solitary or multicentric IM involving the viscera in which surgical resection is not advisable or the lesions are life-threatening, chemotherapy regimens have been described that successfully treated the lesions. These regimens include vinblastine with methotrexate, vincristine alone, vincristine with methotrexate, vincristine with actinomycin-D, and 2-chlorodeoxyadenosine. Caution must be taken to balance the early and late side effects of these agents with their benefits. Complete resection of solitary IM is a reasonable option as long as it is safe to do so. If there are any concerns about possible local complications of resection, then a strategy of careful observation is advisable.
The present case, although it shares common characteristics with previously reported cases of multicentric IM, is unique in several ways. Presentation resulted from a pathologic fracture, whereas typically it is skin or soft-tissue involvement. Also, there was only evidence of osseous involvement, even after extensive investigation. Finally, there was no craniofacial involvement, which has been found to be common in multicentric IM.
Reference: McCammon J, Stefanovici C, Martin RK, Larouche P. Multiple bone lesions in an 8-month-old child presenting with pathologic fracture: a rare case of solely osseous multicentric infantile myofibromatosis. JBJS Case Connect. 2016 Apr-Jun;6(2):e42.
What is the diagnosis?
Polyostotic fibrous dysplasia
Infantile myofibromatosis
Multiple hemangiomas
McCune-Albright syndrome
Possible child abuse