A 5-Year-Old Boy with Painless Neck Stiffness
February 7, 2024
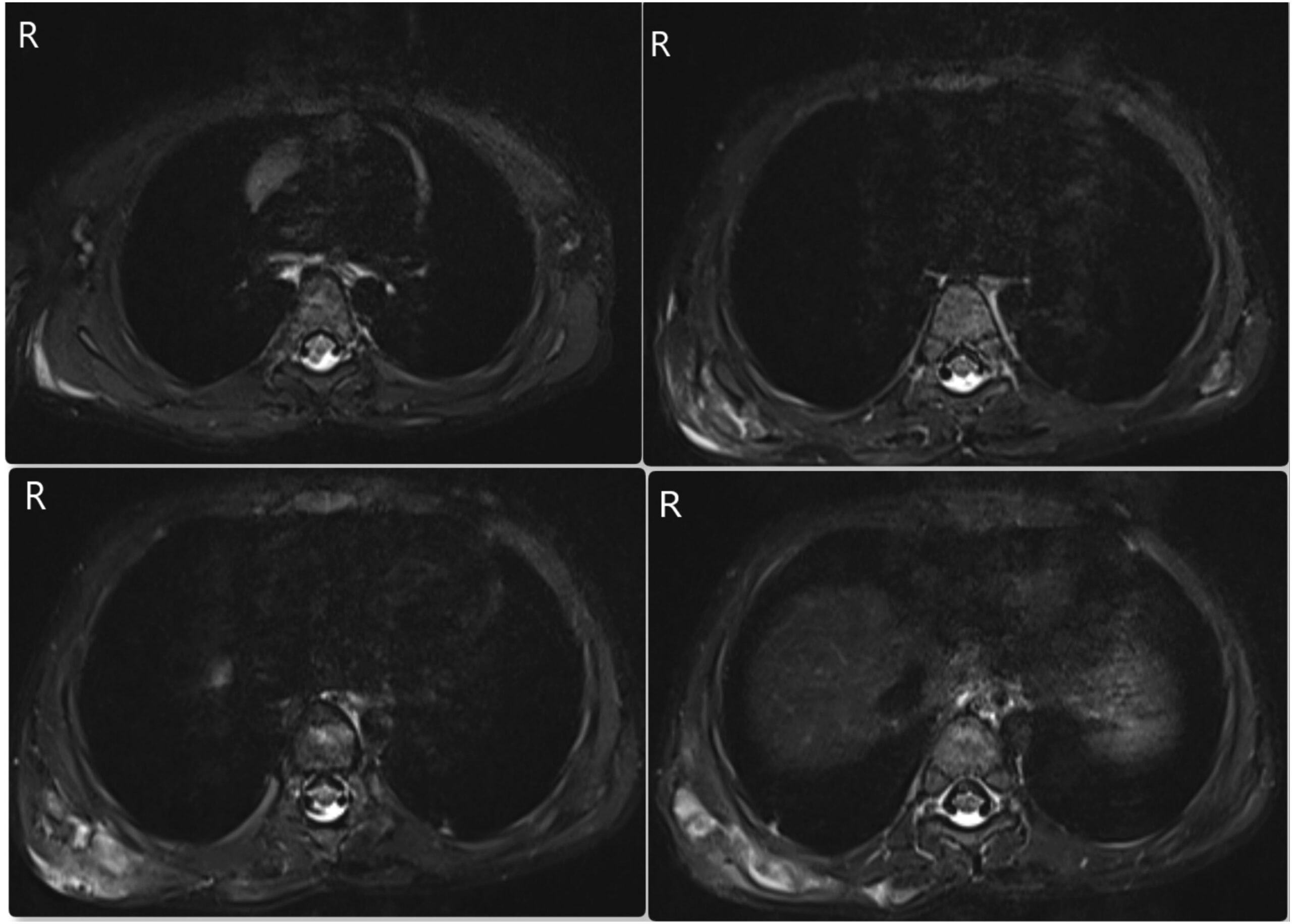
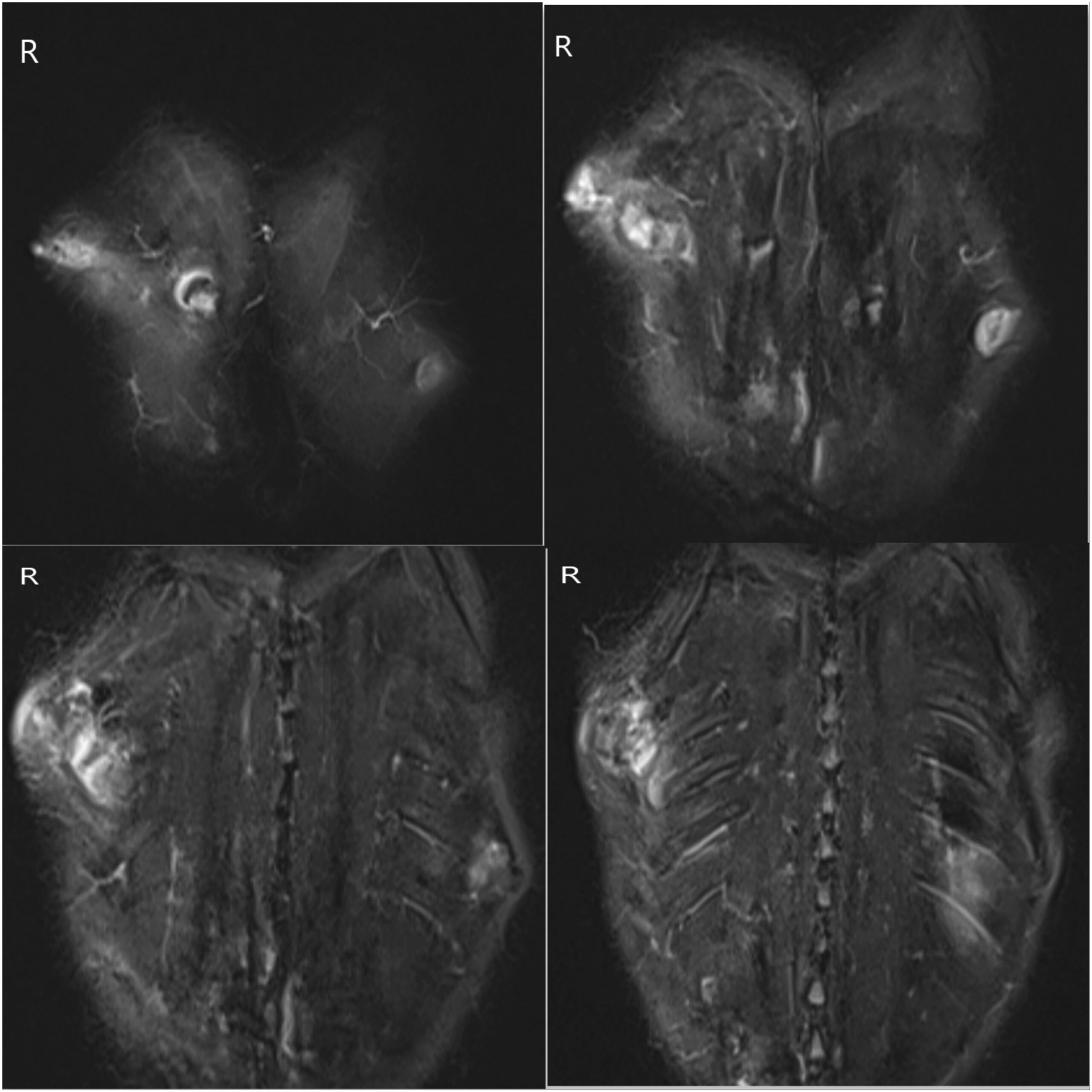
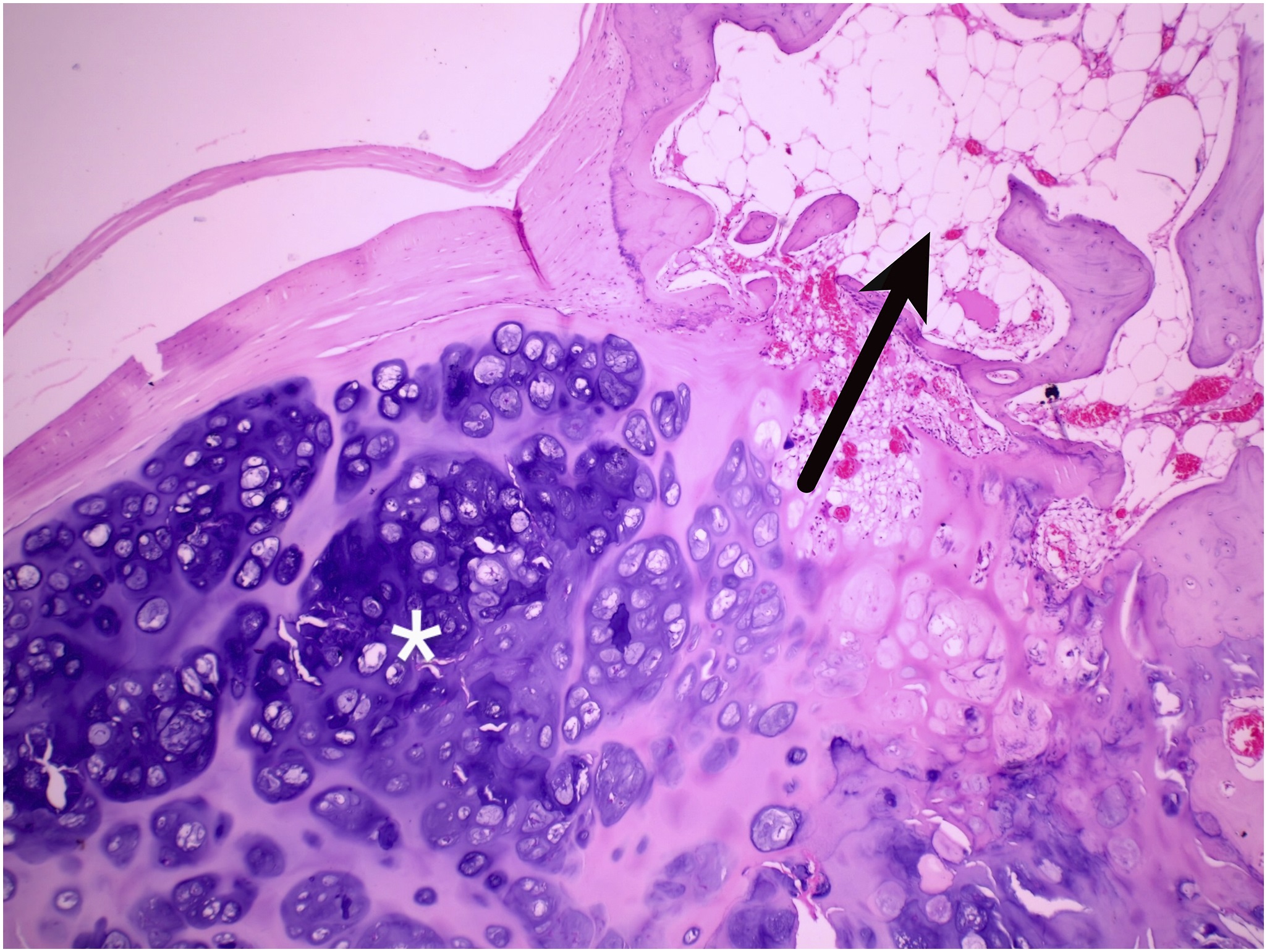
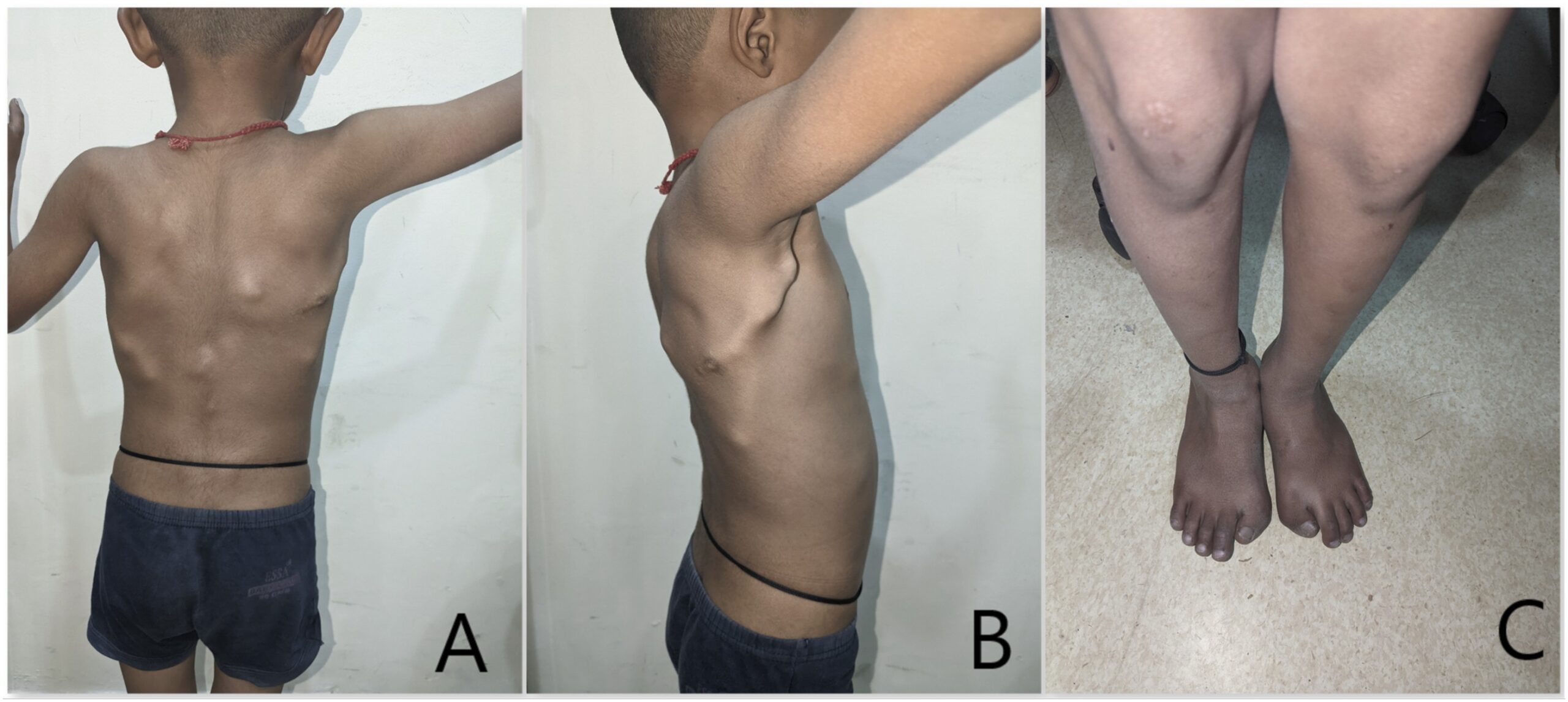
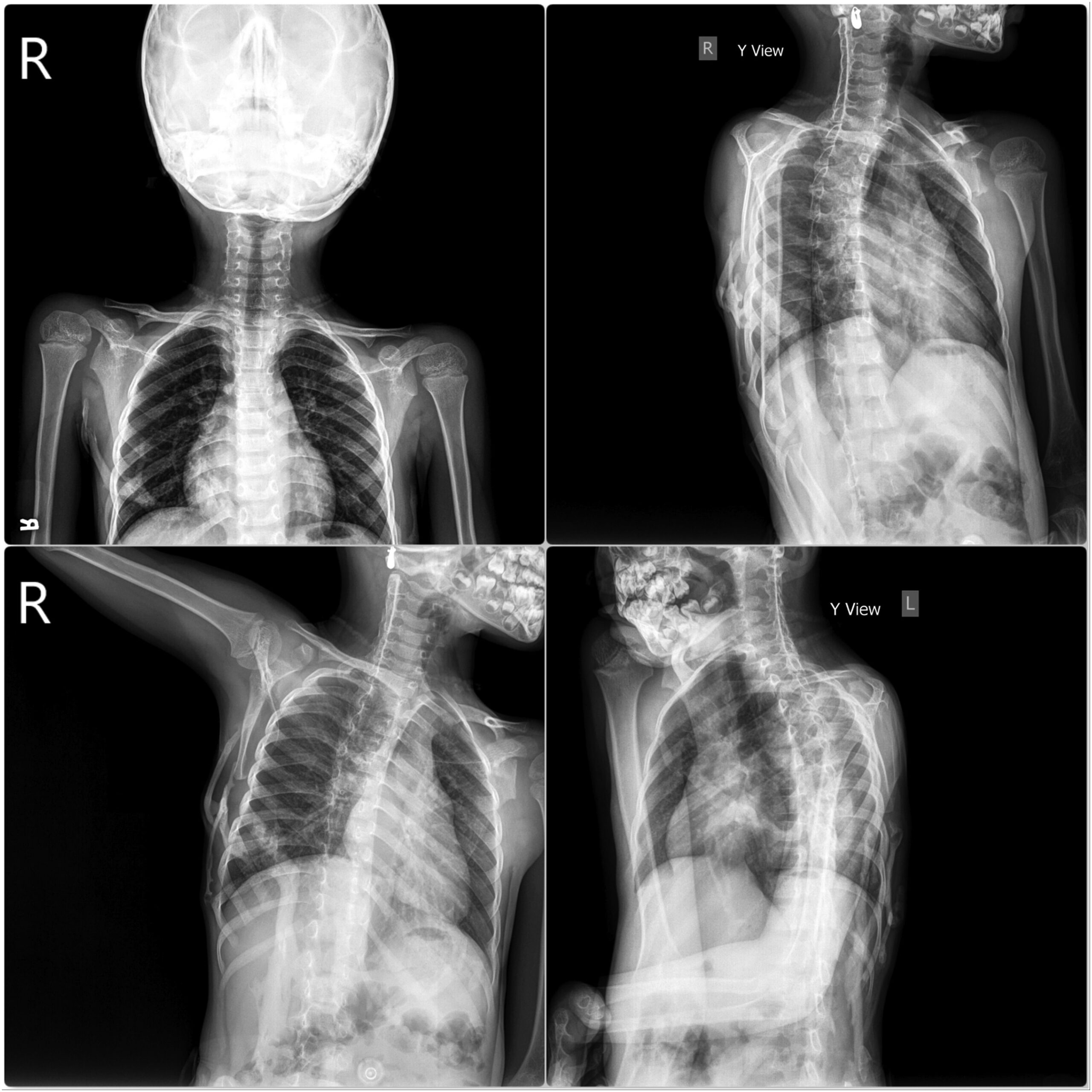
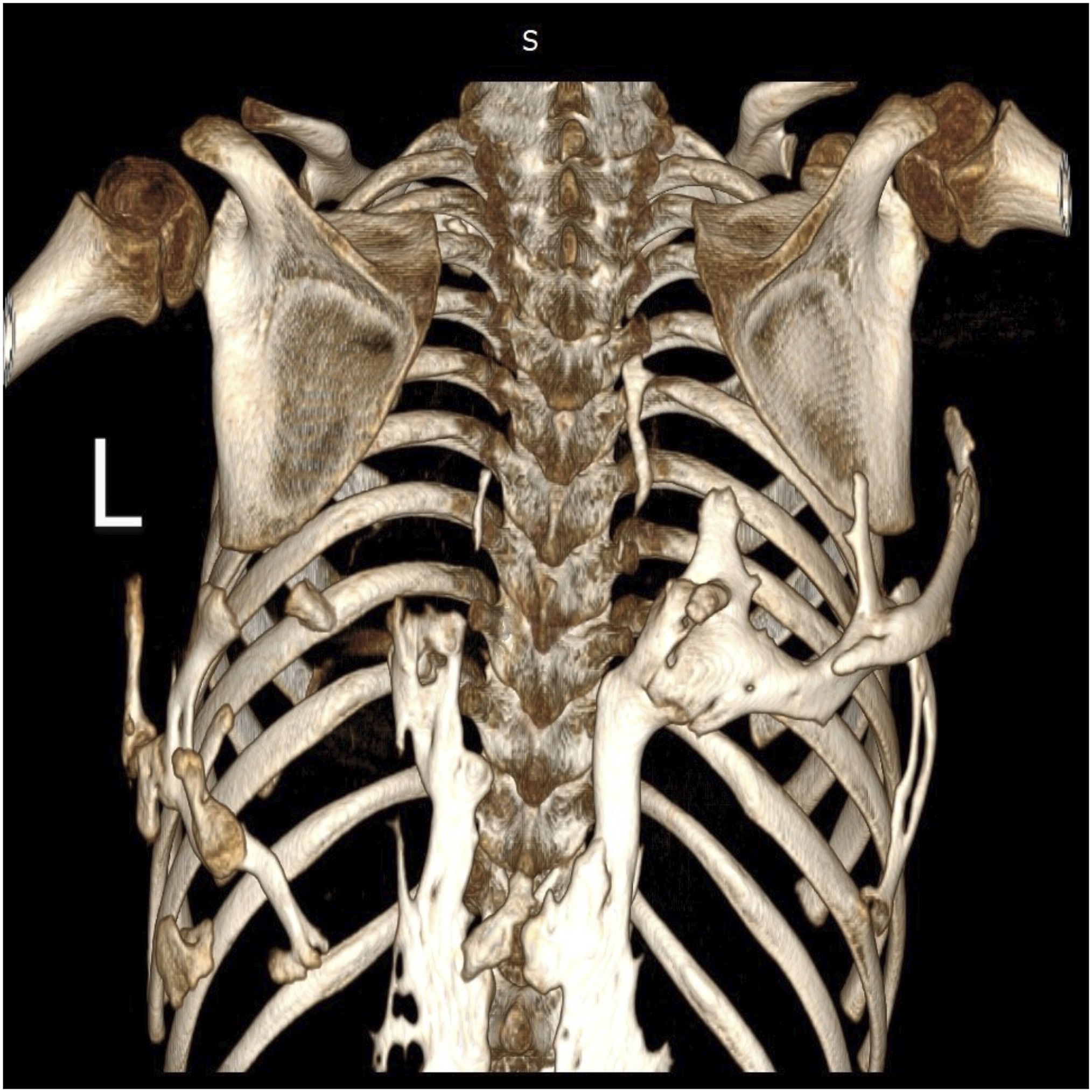
 Fig. 1
Fig. 1 Fig. 2
Fig. 2 Fig. 3
Fig. 3 Fig. 4
Fig. 4 Fig. 5
Fig. 5 Fig. 6
Fig. 6A 5-year-old boy, who had previously been diagnosed with hereditary multiple exostoses, presented with neck stiffness and painless multiple firm immobile masses in the left scapular region. Active and passive movements in the left shoulder were restricted. The flexion and abduction were 60°, internal rotation was 20°, and external rotation was 30°. He did not report any other swellings on his body, waxing or waning of swellings, or hearing loss. He was born full-term vaginally and had an uneventful postnatal period, reaching developmental milestones within the expected time frame.
Two years previously, the patient had presented elsewhere with neck stiffness and swellings over the right scapular region, accompanied by restricted right shoulder movement. Magnetic resonance imaging (MRI) scans at that time revealed multiple ossified lesions, characterized by a cartilaginous cap, along the scapula, ribs, and posterior spinous process (Fig. 1), and MRI scans of the cervical spine revealed a diffuse altered signal intensity along the posterior and lateral neck musculature (Fig. 2). The patient underwent a core needle biopsy, and the result indicated a benign cartilaginous lesion (Fig. 3), leading to a diagnosis of multiple hereditary exostoses. The patient subsequently underwent excision of the right scapular swellings, but developed new bony hard swelling on the right scapular region after the surgical procedure.
During a thorough clinical examination, the patient did not exhibit any syndromic facial features, and the systemic examination did not reveal any notable abnormalities. However, multiple bony hard masses were observed over the upper dorsal spine region. In addition, there were restrictions on neck movements, and the great toes were short and exhibited hallux valgus deformity (Fig. 4).
Radiographs (Fig. 5) and a 3-dimensional (3D) reconstruction of a computed tomographic (CT) scan (Fig. 6) revealed extensive heterotopic bone formation over the dorsal spine region and symmetrical malformation of the great toes. The patient’s laboratory findings were normal.
On the basis of radiographic evidence, the patient was diagnosed with fibrodysplasia ossificans progressiva. Genetic testing identified an ACVR1/ALK2 gene mutation confirming that diagnosis. Genetic testing was not performed on the parents because they were asymptomatic.
The parents of the patient were provided with counseling with regard to the prognosis and the potential for the condition to worsen due to surgical procedures, intramuscular injections, or trauma. Avoiding contact sports, fall prevention measures, and the use of protective headgear were strongly recommended.
Proceed to Discussion >>Reference: K S M, Gupta A. Challenges in diagnosing fibrodysplasia ossificans progressiva: a case report. JBJS Case Connect. 2023 Oct 5;13(4).e23.00327.
Fibrodysplasia ossificans progressiva is caused by an activating mutation in the bone morphogenic protein (BMP) type-I receptor, activin A receptor type 1 (ACVR1), resulting in the misregulation of the BMP signaling pathway. Cohen et al. studied the natural history of fibrodysplasia ossificans progressiva and reported that the neck, spine, and shoulder girdles were the most common sites for heterotopic ossification. Ossification proceeded in the axial to appendicular, cranial to caudal, and proximal to distal directions. Ninety-five percent of patients had restrictive upper-limb mobility by 15 years. Kaplan et al. noted a shortened life span in individuals with fibrodysplasia ossificans progressiva, with a median life span of 56 years (95% confidence interval, 51 to 60 years). The most common cause of death was cardiorespiratory failure from thoracic insufficiency syndrome. In addition to pulmonary complications, fibrodysplasia ossificans progressiva can lead to temporomandibular joint ankylosis, which can result in difficulties in eating and substantially affect quality of life. Conduction hearing is a common phenotypic feature probably due to middle ear ossification. Routine biochemical evaluations are usually normal, although alkaline phosphatase and erythrocyte sedimentation rate may be increased, especially during flare-ups.
Proximal tibial osteochondromas are a common phenotypic feature in fibrodysplasia ossificans progressiva and are also reported in the proximal fibula, distal tibia, distal femur, and coronoid process of the mandible. They are usually pedunculated and symmetrical, lack a bulbous tip, are pointed away from the joint, and rarely cause bursitis. Excision can cause exuberant ossification. Hereditary multiple exostoses are an autosomal dominant disorder characterized by the mutation of EXT1 and EXT2 genes associated with multiple osteochondromas, which are usually sessile, are commonly found around the knee, and can arise from metaphysis or diaphysis of long bones. Those that are pedunculated have a narrow stalk with a bulbous tip, pointed away from the joint. Bursitis, neuropathy, and malignant transformation (common in the axial skeleton) are common reasons for the excision. Angular leg deformities, hand and forearm deformities (Madelung deformity), and short stature are additional features.
Histopathology of both fibrodysplasia ossificans progressiva and hereditary multiple exostoses would reveal osteochondral tissue because it is caused by defective induction of endochondral osteogenesis. Osteochondroma formation in both conditions is mediated by disruption of the BMP/Ihh/PTHrP negative feedback loop. In fibrodysplasia ossificans progressiva, this feedback loop is affected directly through the increased activity of mutated ACVR1, whereas in hereditary multiple exostoses, the feedback loop is affected indirectly through a reduction in heparan sulphate proteoglycans, leading to an increase of BMP and Ihh morphogens that regulate BMP/Ihh/PTHrP signaling.
Fibrodysplasia ossificans progressiva can be differentiated from hereditary multiple exostoses with the typical deformities, location, and morphology of exostoses.
Due to its characteristic tender, rubbery, soft-tissue induration with rapid progression in size, before the appearance of heterotopic ossification, fibrodysplasia ossificans progressiva is often mistaken for juvenile aggressive fibromatosis (desmoid tumor) or soft-tissue sarcoma. Juvenile aggressive fibromatoses are locally invasive neoplasms with initial rapid growth and development of contractures restricting joint mobility. Fibromatoses usually appear in the extremities, followed by the trunk and the head-neck region. It tends to spontaneous regression and has a propensity for local recurrence. Juvenile fibromatoses can be familial or sporadic and may be associated with familial adenomatous polyposis and Gardner syndrome.
Soft-tissue sarcomas typically manifest as solitary, constantly growing painless masses with a soft to firm consistency. MRI scans would reveal a heterogeneous mass, but confirmation requires a biopsy. By contrast, fibrodysplasia ossificans progressiva lesions abruptly appear and subsequently undergo rapid and dynamic changes in size and shape, often occurring within a matter of hours, far greater than the rate of change seen in most tumors.
Sarcomas and fibromatoses have spontaneous onset, unlike fibrodysplasia ossificans progressiva masses, which are triggered by trauma. The combination of malformed great toes and axial soft-tissue masses should raise the suspicion of fibrodysplasia ossificans progressiva. A genetic test would confirm the diagnosis before proceeding with invasive procedures.
Other important differential diagnoses to consider are conditions associated with heterotopic ossification. Major genetic disorders with heterotopic ossification are fibrodysplasia ossificans progressiva, Albright hereditary osteodystrophy, and progressive osseous heteroplasia. Albright hereditary osteodystrophy and progressive osseous heteroplasia are characterized by a mutation in the GNAS gene. Albright hereditary osteodystrophy presents ectopic dermal and subcutaneous tissue ossification, brachydactyly, short stature, and a round face. Albright hereditary osteodystrophy characteristically presents with intramembranous ossification in contrast to fibrodysplasia ossificans progressiva, in which enchondral ossification is seen. Progressive osseous heteroplasia is extremely rare, and patients are normal at birth. Ossification appears in the dermis in a dermatome distribution and spreads into deeper tissue in a characteristic reticular pattern leading to joint ankylosis.
Metachondromatosis is associated with both osteochondroma and enchondroma formation. It is associated with the mutation of the PTPN11 gene. Lesions are oriented away from the epiphysis and have a predilection for hands and feet. These lesions rarely undergo malignant transformation, and spontaneous regression is expected.
In conclusion, diagnosis of fibrodysplasia ossificans progressiva is often overlooked or delayed, thus resulting in possible inopportune testing (including biopsies and other surgical interventions), which could worsen the clinical course and lead to poor patient outcomes. Orthopaedic surgeons should keep all differential diagnoses in mind and perform thorough clinical examinations, looking for often subtle clinical clues while diagnosing these conditions.
Reference: K S M, Gupta A. Challenges in diagnosing fibrodysplasia ossificans progressiva: a case report. JBJS Case Connect. 2023 Oct 5;13(4).e23.00327.
What is the diagnosis?
Hereditary multiple osteochondromas
Chondrosarcoma arising in the cap of an osteochondroma
Metachondromatosis
Fibrodysplasia ossificans progressiva
Albright hereditary osteodystrophy