A Nineteen-Year-Old Woman with a Swollen Finger
November 16, 2011

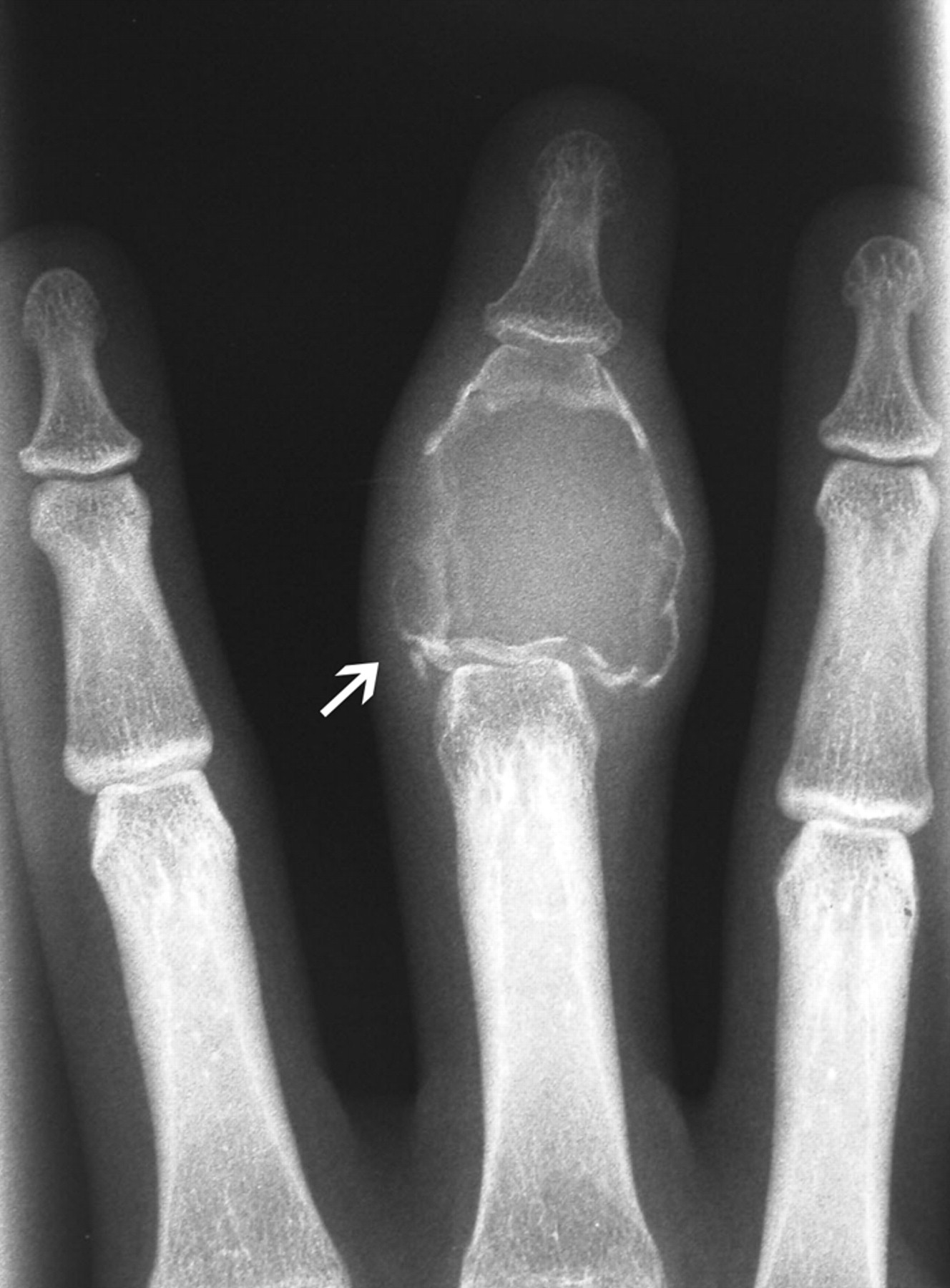

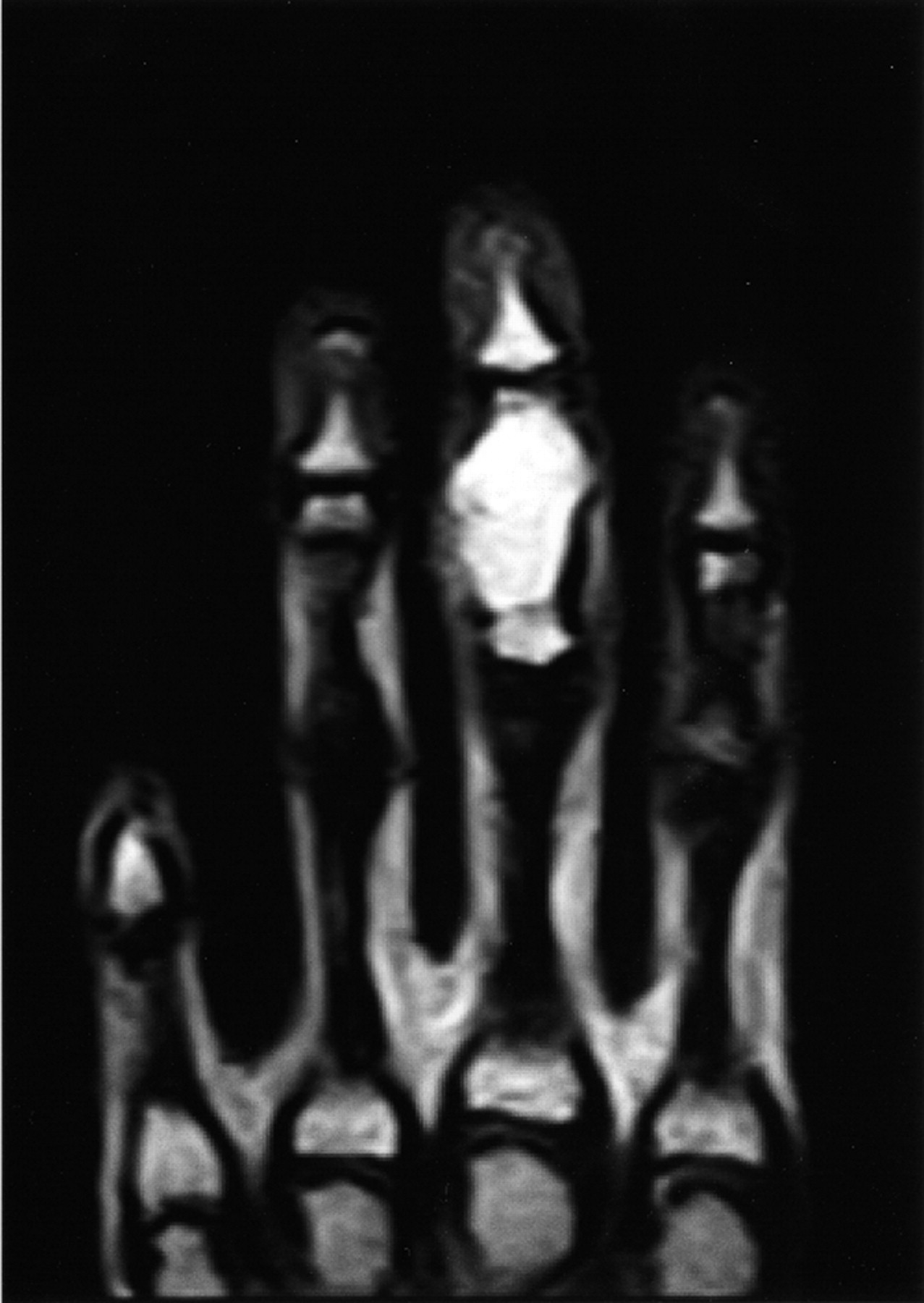
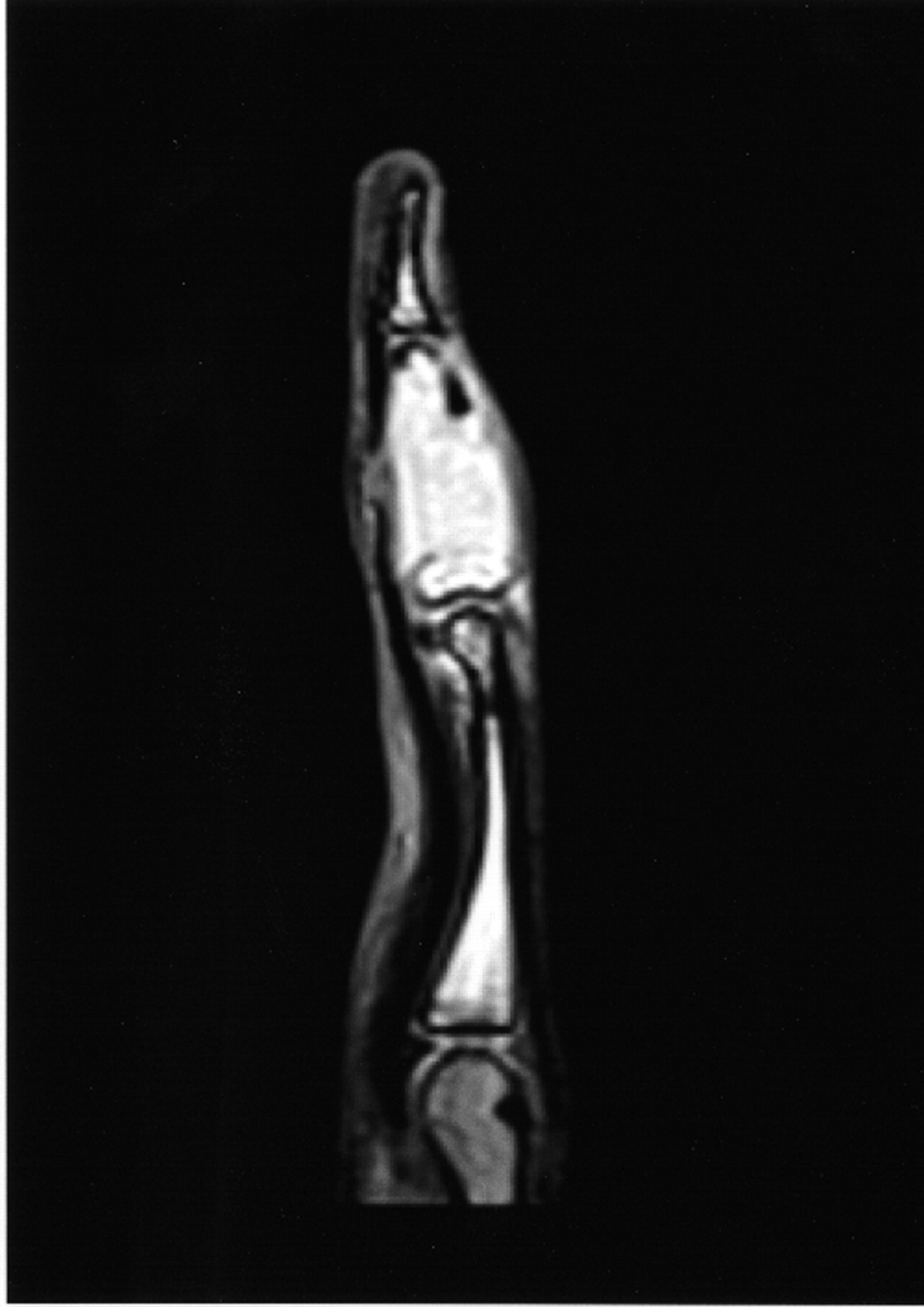
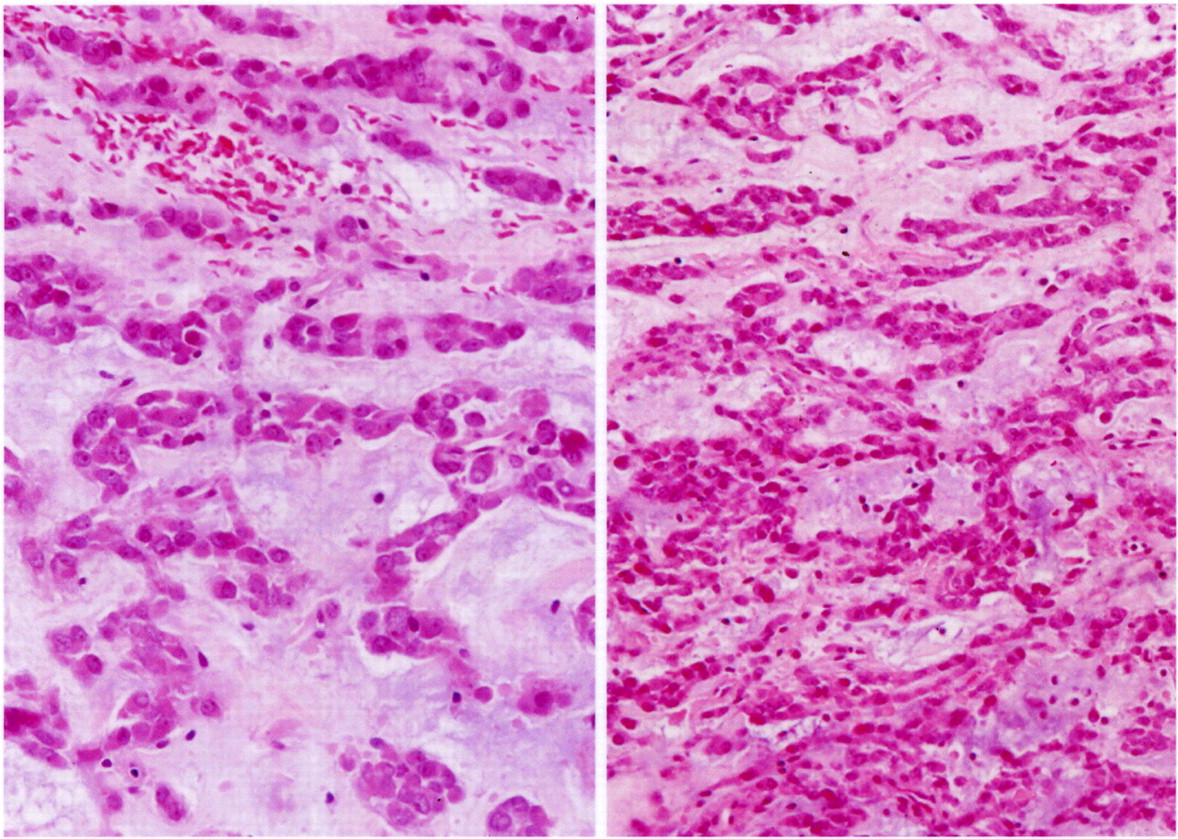

 Fig. 1
Fig. 1 Fig. 2-A
Fig. 2-A Fig. 2-B
Fig. 2-B Fig. 3-A
Fig. 3-A Fig. 3-B
Fig. 3-B Fig. 4
Fig. 4 Fig. 5
Fig. 5 Fig. 4
Fig. 4 Fig. 5
Fig. 5 Fig. 6
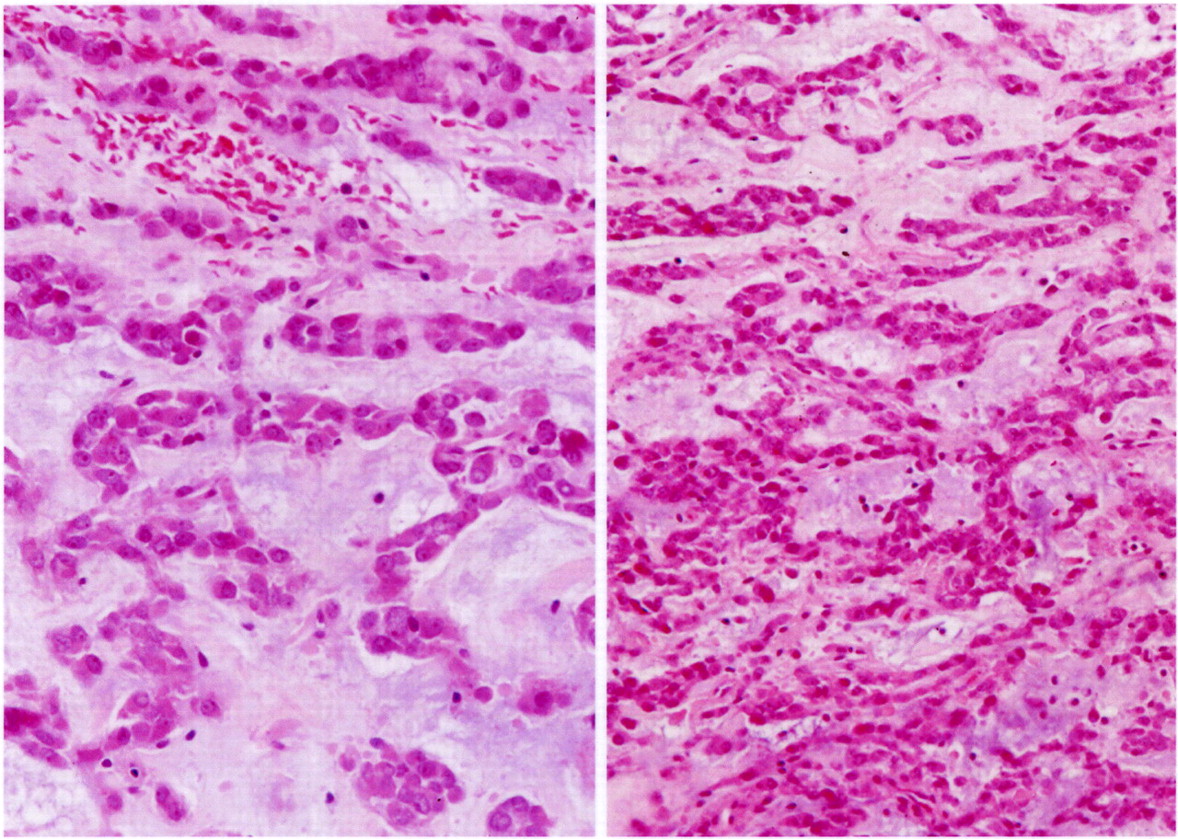
Fig. 6A nineteen-year-old woman presented with a nine-month history of a progressively enlarging, painful mass of the right long finger. There was no history of trauma or foreign-body penetration. She had no constitutional symptoms, and the medical history was remarkable only for asthma. On examination, there was a 2 × 1.5 × 1.5-cm mass involving the middle phalanx (Fig. 1). There was decreased active digital flexion, mild ulnar deviation of the finger, and normal sensation. The patient had no antecubital or axillary lymphadenopathy. Radiographs demonstrated a lytic expansile lesion of the middle phalanx of the long finger (Figs. 2-A and 2-B). No full-thickness cortical destruction was evident, although the extensive endosteal scalloping made this difficult to confirm. The lesion extended to the proximal articular surface, where a pathologic fracture was noted. The proximal and distal interphalangeal joints were unaffected. No matrix formation was visible. Magnetic resonance imaging (MRI) studies showed a marrow-replacing lesion filling the entire middle phalanx that was hyperintense compared with skeletal muscle on water-sensitive images (Figs. 3-A and 3-B). No soft-tissue extension was visible. On the basis of the benign radiographic assessment, an excisional biopsy was planned. At surgery, there was extensive bone destruction and only a thin, incomplete portion of dorsal cortex and several small areas of articular cartilage at the proximal and distal interphalangeal joints were intact. We performed an intralesional excision with attempted preservation of the articular surfaces and as much of the middle phalanx as possible. No soft-tissue component was evident. A cortical strut graft together with cancellous bone graft harvested from a clean surgical field of the distal end of the radius was used to reconstruct the middle phalanx. Grossly, the excised tissue had a lobulated, gelatinous consistency without evident calcification. The histological photographs are shown in Figs. 4 and 5.
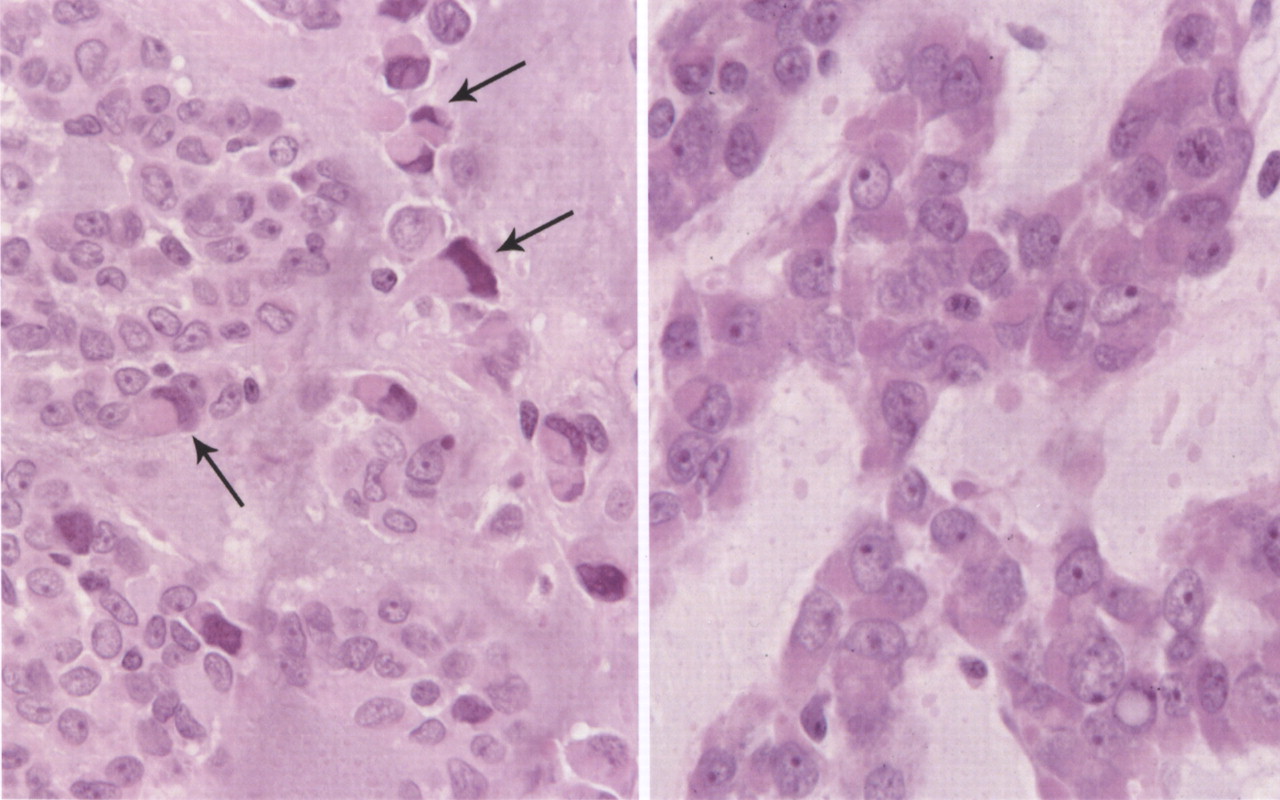
Histologically, the tumor was composed of small epithelioid-like cells, with densely eosinophilic cytoplasm, arranged in strands, cords, and small nests within an abundant blue-gray myxoid stroma (Fig. 4). In most of the tumor, the cells had mildly atypical nuclei with a uniform chromatin pattern. However, in some foci, the nuclei were enlarged with moderate atypia and prominent nucleoli. Scattered cells containing densely eosinophilic globular cytoplasmic inclusions that distorted and compressed the nuclei, creating the appearance of rhabdoid cells, were also found (Fig. 5). Mitotic figures were noted, with as many as four per high-powered field. No evidence of cartilage, chondroid tissue, or cells in lacunar spaces was found. Histochemical studies showed the myxoid stroma to be strongly stained by alcian blue at both pH 1.0 and 2.5, with the staining being resistant to change by pretreatment of the tissue sections with bovine testicular hyaluronidase. The stroma also yielded a metachromatic reaction with toluidine blue. Immunostaining showed diffuse and strong tumor cell reactivity for vimentin, neuron-specific enolase, chromogranin, and synaptophysin, with retention of nuclear reactivity for the INI-1 gene product with use of BAF47 antibody. Immunostaining for S-100 protein and epithelial membrane antigen were only weakly and focally positive, while that for cytokeratin (AE1/AE3) was negative. With use of paraffin sections of formalin-fixed tumor tissue, a fluorescence in situ hybridization (FISH) study with a commercial Ewing sarcoma (EWS, 22q12) break-apart probe (Vysis, Downers Grove, Illinois) showed widely separated red and green signals found in two-thirds of 200 counted interphase tumor cells (Fig. 6). This finding is consistent with the presence of the EWS-containing chromosomal translocation typically seen in extraskeletal myxoid chondrosarcoma. On the basis of the results of the various tissue studies, a diagnosis of skeletal myxoid chondrosarcoma was made. After the diagnosis, a computed tomography scan of the chest and a bone scan were performed to evaluate for metastatic disease. Both were negative. The patient underwent a ray resection of the long finger without transposition seventeen days after the initial procedure. Histologic analysis of the amputation specimen showed only a solitary microfocus of a few residual tumor cells within the periosteal soft tissue adjacent to the middle phalanx; resection margins were otherwise free of tumor. One year postoperatively, the patient was healthy with an aesthetically acceptable hand and full finger flexion.
Proceed to Discussion >>Reference: Bumpass, DB, Kyriakos M, Rubin DA, Manske PR, Goldfarb CA. Myxoid chondrosarcoma of the phalanx with an EWS translocation. A case report and review of the literature. J Bone Joint Surg Am. 2011;93:e23(1-7).
On review of the English-language literature, we found forty-six possible cases of skeletal myxoid chondrosarcoma. Thirty-two cases fit our inclusion criteria of clear intraosseous origin with reliable pathologic and radiographic diagnosis. Cases were eliminated if they were of periosteal origin, had a possible soft-tissue origin, had an unclear histologic diagnosis, were not clinically detailed or illustrated, or were reported more than once. Of the thirty-two cases of skeletal myxoid chondrosarcoma that were included, sixteen were in male patients, fourteen were in female patients, and two had no data on sex. The age at the time of diagnosis ranged from nine to seventy years, with a mean of forty-two years. The femur was the most common site of involvement, accounting for nine cases (28%). Six tumors (19%) occurred in the pelvis, and five tumors (16%) occurred in the foot. Only one case (3%) occurred in the hand. Immunostaining results were reported for only five previous cases. Tumor cell reactivity for S-100 was positive in five of five cases (100%) but with variable intensity, and vimentin was strongly positive in two of two cases (100%). Cytokeratin and epithelial membrane antigen were absent in all tumors tested. Treatment was described for twenty-nine of the thirty-two patients. Surgical excision, ranging from local curettage to limb amputation, was performed in all twenty-nine patients. Four patients also received radiation therapy and four, chemotherapy. Follow-up data were available for twenty-six patients at a mean of forty-nine months (range, six to 148 months) after diagnosis. Local recurrence developed in eleven patients (42%). Metastases developed in eight patients (31%), six of whom also had local recurrences. The lung was the most common site of metastasis, seen in six of the eight patients. Four patients (15%), three of whom had confirmed metastases, died of the disease. Only three cases of myxoid chondrosarcoma in the hand or wrist have been reported with clinical details, and all were extraskeletal. All three patients were treated with ray resection and were disease-free at the time of the latest evaluation. Wide surgical resection remains the most accepted treatment; little success has been documented with use of chemotherapy and radiation in controlling myxoid chondrosarcoma tumors. Myxoid chondrosarcoma is a distinct subtype of chondrosarcoma with several key differences from conventional chondrosarcoma of bone. First, myxoid chondrosarcoma tumors do not contain substantial amounts of hyaline cartilage or mineralized matrix, traits characteristic of conventional chondrosarcoma. Rather, myxoid chondrosarcoma is composed of cords and strands of small eosinophilic cells within an extensive myxoid stroma, with a lace-like configuration. However, myxoid chondrosarcoma can also contain highly compact, cellular zones lacking a myxoid stroma; foci of extensively pleomorphic and anaplastic cells; and, as in our patient, rhabdoid cells. Second, myxoid chondrosarcoma has characteristic immunohistochemical properties. The myxoid stroma of myxoid chondrosarcoma contains sulfated acid mucopolysaccharides, demonstrated by strong alcian blue staining resistant to hyaluronidase pretreatment, and by metachromatic staining with toluidine blue. Extraskeletal myxoid chondrosarcoma tumors have shown strong reactivity for vimentin in almost all tumors studied. Reactivity for S-100 protein, found in almost all true cartilaginous neoplasms, is positive only in a minority of patients with myxoid chondrosarcoma. In our review of previous series of extraskeletal myxoid chondrosarcoma, only seventy-two (43%) of 169 tumors were positive for S-100, and this reactivity was typically focal and weak. Also, immunostaining for epithelial differentiation was rarely positive; only fourteen (11%) of 123 tumors showed epithelial membrane antigen reactivity, and just four (3%) of 157 were reactive for cytokeratin. Cases of myxoid chondrosarcoma have also been reported with positive staining for neuron-specific enolase, chromogranin, and synaptophysin; all of these are characteristic of tumors with a neuroendocrine lineage and suggest a separate histogenesis from conventional chondrosarcoma. In our patient, the immunostaining results showed strong and diffuse reactivity for vimentin, neuron-specific enolase, chromogranin, and synaptophysin; weak and focal reactivity for S-100 protein; and absent epithelial membrane antigen and cytokeratin reactivity. This is entirely consistent with a diagnosis of myxoid chondrosarcoma. Third, myxoid chondrosarcoma has unique genetic markers that differentiate it from conventional chondrosarcoma. Extraskeletal myxoid chondrosarcoma tumors demonstrate one of several nonrandom reciprocal chromosomal translocations in at least 80% of cases. The most common of these, t(9;22)(q22-31;q12), accounts for approximately 75% of the positive cases and fuses the EWS gene (22q12) with the CHN gene (9q22, also called TEC, NOR1, or MINOR). The hybridization method used in our patient established the presence of the EWS-containing chromosomal translocation (22q12) but did not indicate the associated fusion partner. Although the EWS translocation is also found in a variety of other tumor types, including Ewing sarcoma-primitive neuroectodermal tumor, clear cell sarcoma, desmoplastic small-cell tumor, myxoid liposarcoma, and angiomatoid fibrous histiocytoma, these are easily distinguished from myxoid chondrosarcoma by their routine histomorphologic and histochemical characteristics. In our patient, the EWS translocation is consistent with the most common translocation seen in cases of extraskeletal myxoid chondrosarcoma, t(9;22) EWS-CHN. Despite the histologic similarity between extraskeletal and skeletal myxoid chondrosarcoma tumors, some authors believe them to be separate entities primarily because of the absence of a chromosomal translocation in the six cases of skeletal myxoid chondrosarcoma examined. Rather, skeletal myxoid chondrosarcoma has been viewed as a variant of conventional chondrosarcoma with prominent myxoid degeneration. One possible, and controversial, exception to this involves the report by Gill et al. of a myxoid chondrosarcoma tumor that demonstrated the t(9;22) translocation involving the scapula of a sixty-six-year-old man. On the basis of similar clinical details and a shared source institution, we believe that this is the same patient described by Sciot et al. and by Kilpatrick et al. Because of the reported marked destruction of the scapula with an extensive soft-tissue component in this case, we believe, as do others, that there is doubt concerning the origin of this particular tumor in bone versus soft tissue. Unfortunately, no radiographs were provided. Thus, prior to the case of our patient, no definitive intraosseous myxoid chondrosarcoma tumor has been described with the EWS translocation. We considered the possibility that the tumor in our patient might represent a metastasis from an extraskeletal primary myxoid chondrosarcoma tumor. In 2007, Ehara et al. documented the cases of four patients in whom soft-tissue myxoid chondrosarcoma tumors had metastasized to bone, and these metastases were both local and distant. However, we did not identify a soft-tissue primary tumor on physical examination or on subsequent metastatic work-up. Also, although extraskeletal myxoid chondrosarcoma can secondarily involve bone by direct extension, the tumor in our case clearly originated within bone, on the basis of the operative, radiographic, and pathologic findings. Extensive myxoid degeneration in an otherwise conventional osseous chondrosarcoma may yield biopsy tissue that is indistinguishable from that of true skeletal myxoid chondrosarcoma. However, the usual radiographic pattern of abundant cartilaginous matrix with calcification in conventional chondrosarcoma is in contrast to the osteolytic pattern typically seen in myxoid chondrosarcoma. Additionally, conventional chondrosarcoma with myxoid change contains abundant malignant-appearing hyaline cartilage, has strong and diffuse S-100 protein reactivity, lacks neuroendocrine differentiation, and possesses no characteristic chromosomal translocations. Our difficulty in accurately diagnosing this tumor prior to initial resection was based on the relatively nonspecific appearance of the lesion on radiographs and magnetic resonance imaging (MRI) and the rarity of all types of chondrosarcoma in the finger phalanges. In retrospect, rather than an intralesional excision, an incisional biopsy with staged definitive treatment would have been ideal. This difficulty in preoperative diagnosis was also encountered by Kwon et al., who performed a needle biopsy of a calcaneal myxoid chondrosarcoma after MRI led to a presumptive diagnosis of benign chondromyxoid fibroma. While the imaging findings in our patient were consistent with previous descriptions of skeletal myxoid chondrosarcoma—namely, a lytic lesion without internal calcifications and with cortical expansion and endosteal scalloping—many of these features also characterize finger enchondromas. With MRI, skeletal myxoid chondrosarcoma demonstrates a lobulated appearance with intense enhancement after injection of gadolinium contrast medium. In the absence of contrast, T2 signal within the tumor is more intense than T1 signal. To our knowledge, the case of our patient is the first definitive example of a primary intraosseous myxoid chondrosarcoma tumor containing the same chromosomal translocation as found in its extraosseous counterpart. On the basis of this case, we believe there is no essential difference between skeletal and extraskeletal myxoid chondrosarcoma tumors in terms of their cytomorphology, immunohistochemical features, and genetic character, and they should be considered as a single tumor type.
Reference: Bumpass, DB, Kyriakos M, Rubin DA, Manske PR, Goldfarb CA. Myxoid chondrosarcoma of the phalanx with an EWS translocation. A case report and review of the literature. J Bone Joint Surg Am. 2011;93:e23(1-7).

Myxoid chondrosarcoma
Digital myxoid cyst
Chondromyxoid fibroma
Myxoid liposarcoma