A Sixty-three-Year-Old Woman with Right Hand and Arm Deformity
May 15, 2013
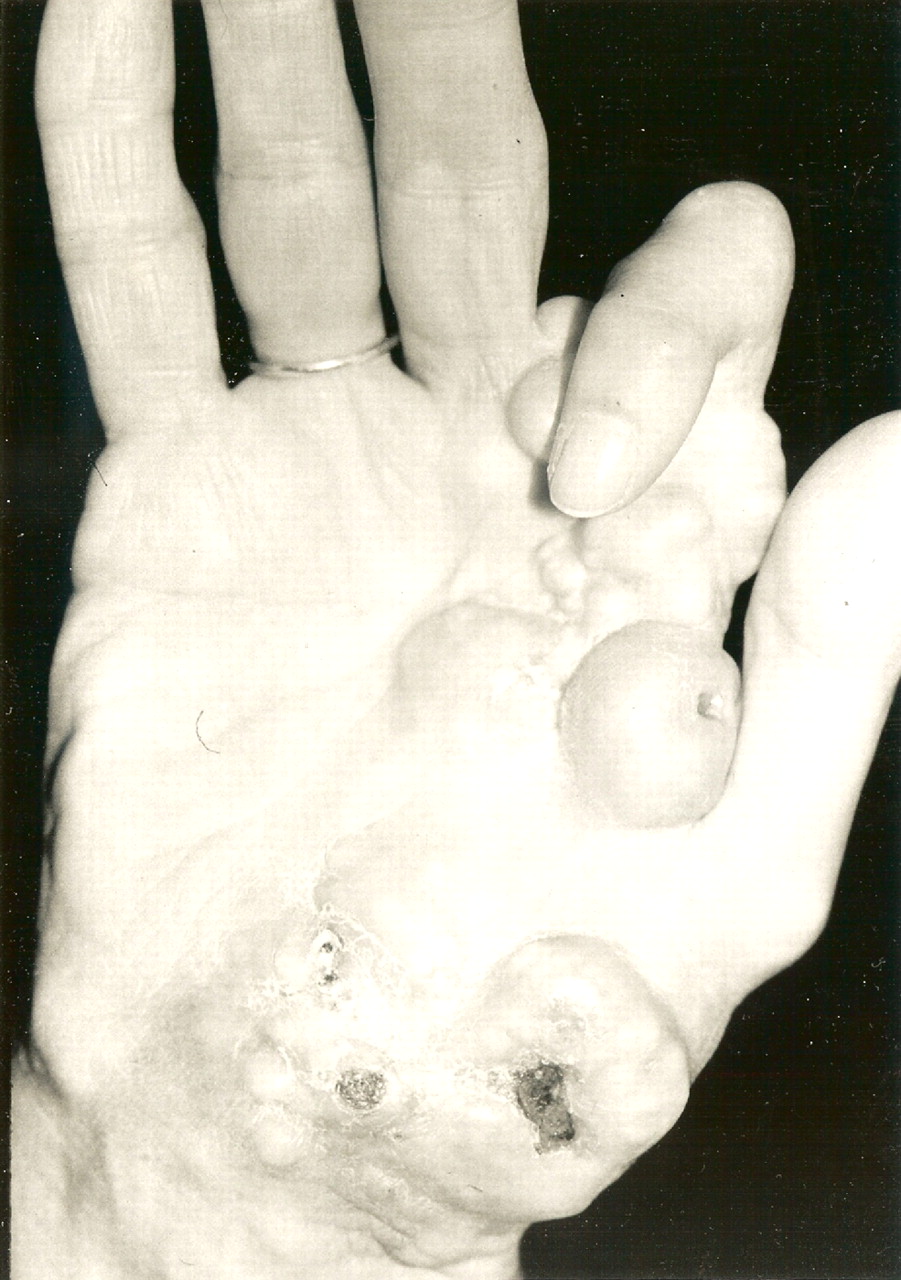
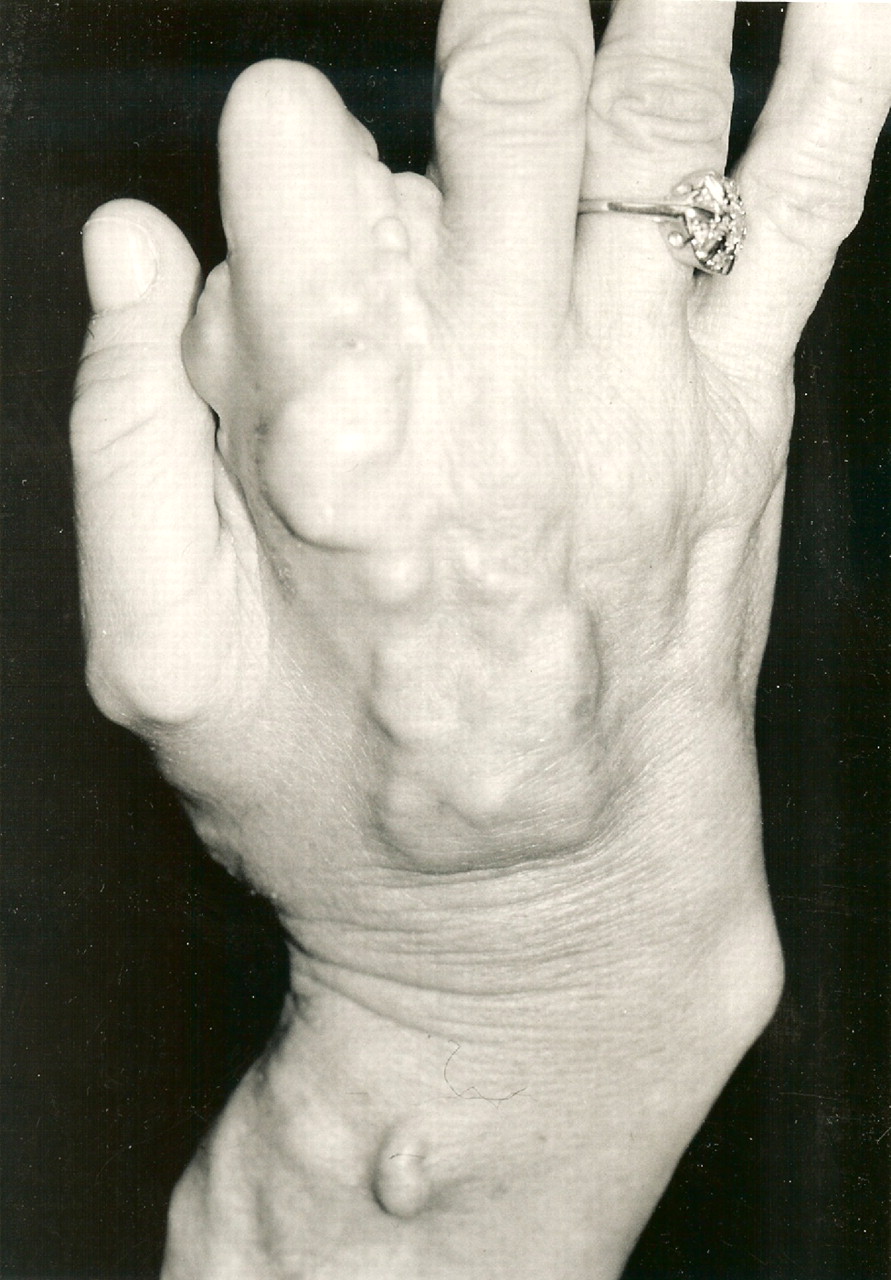
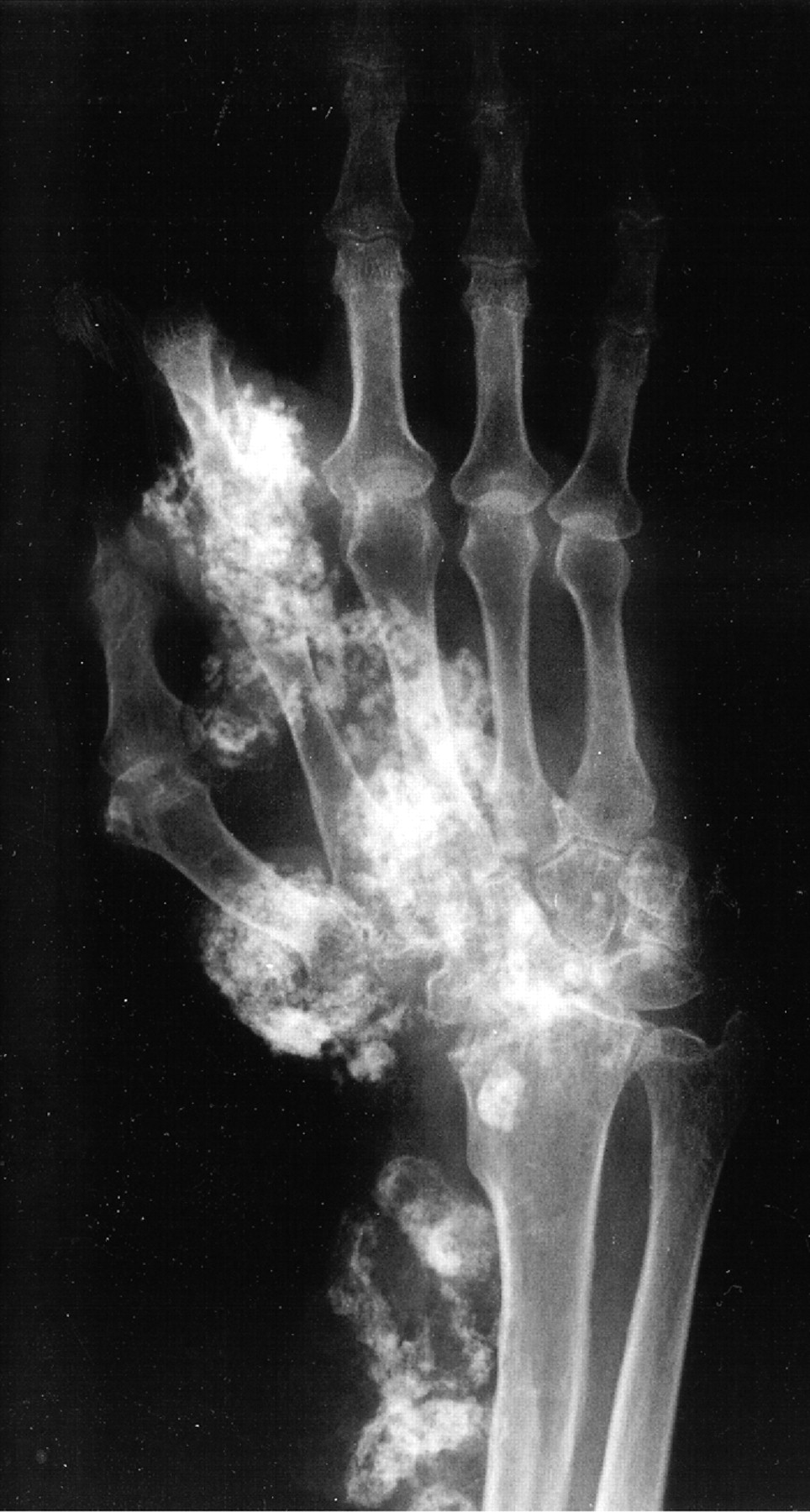
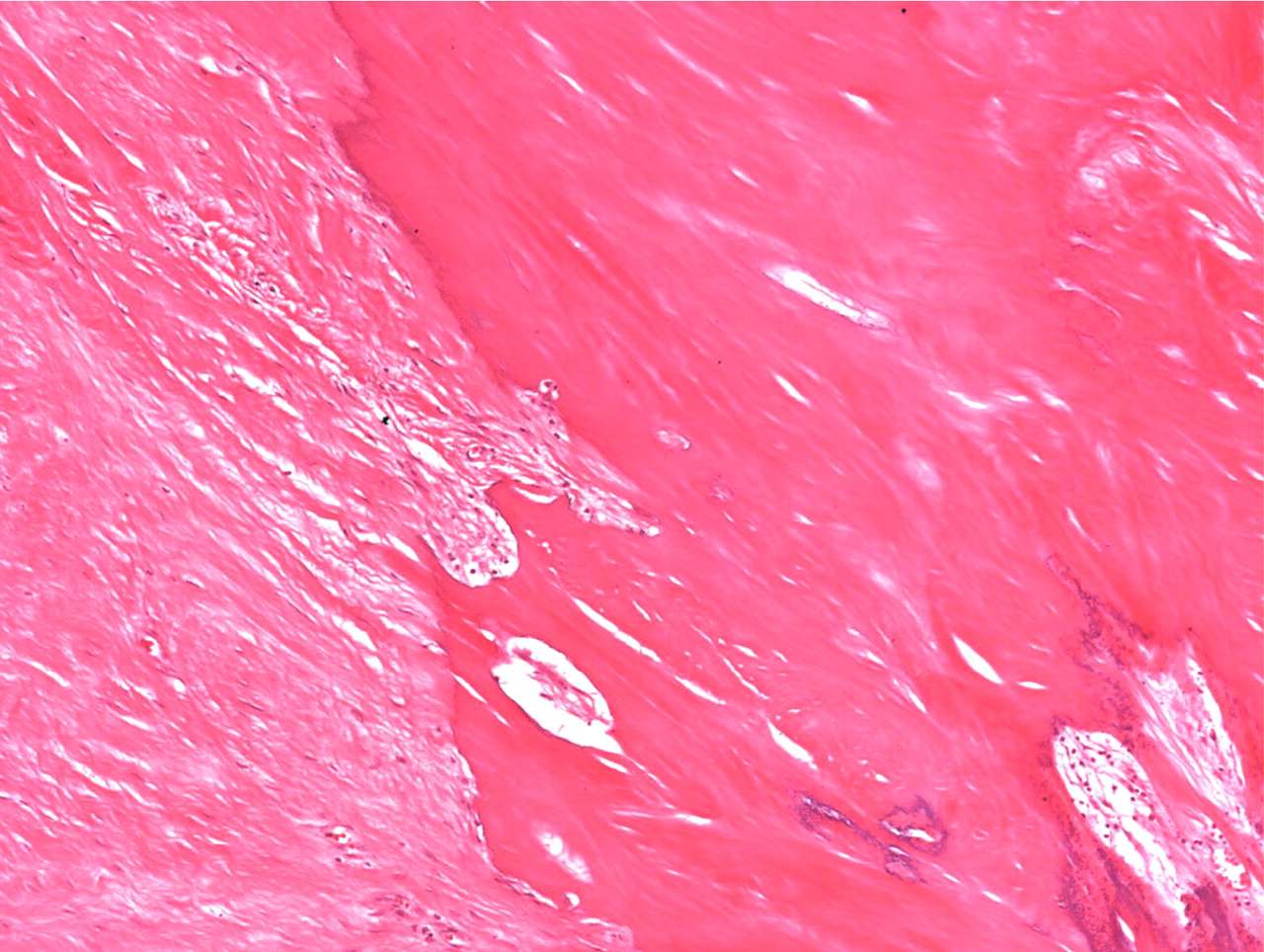

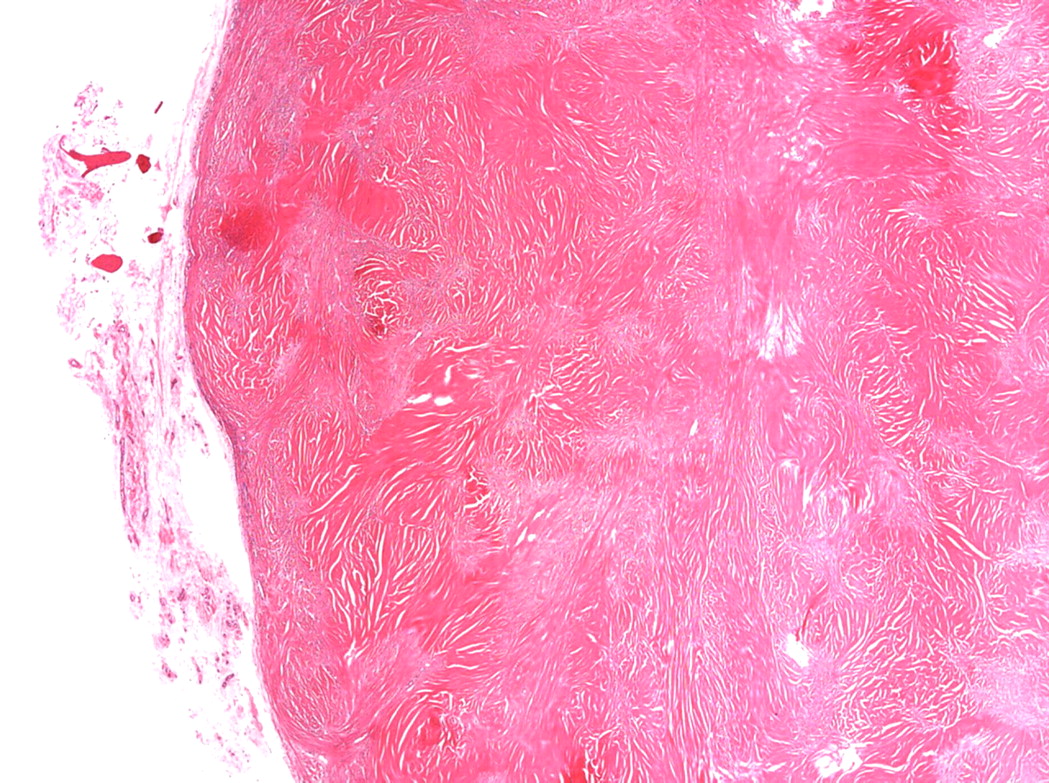
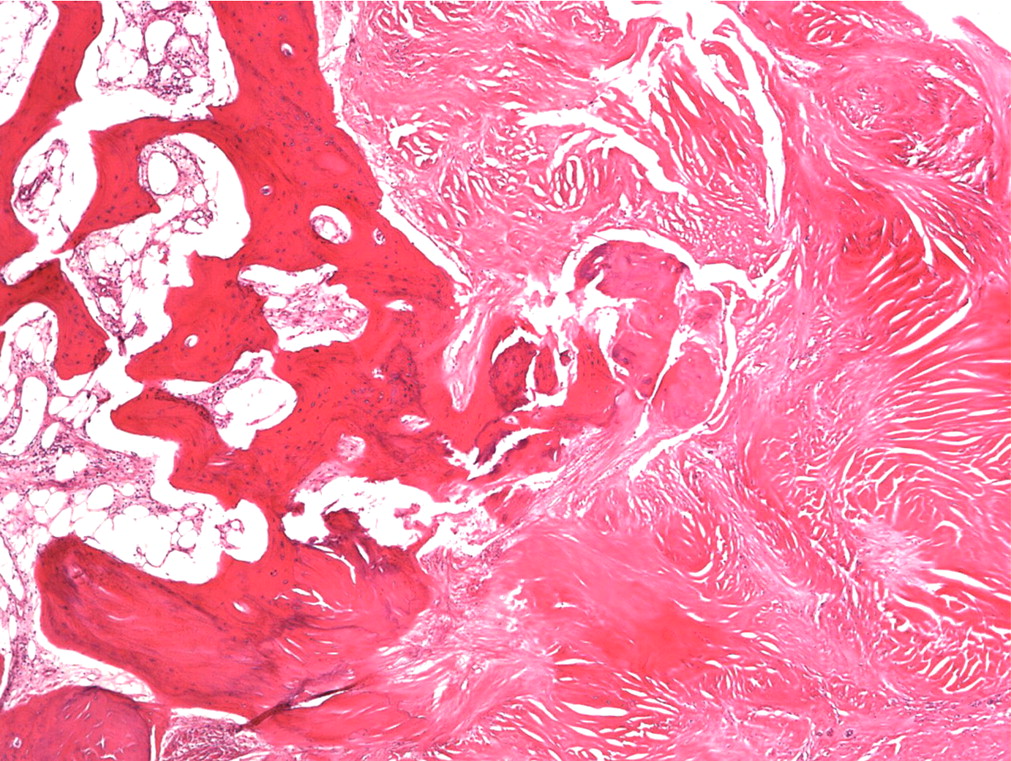
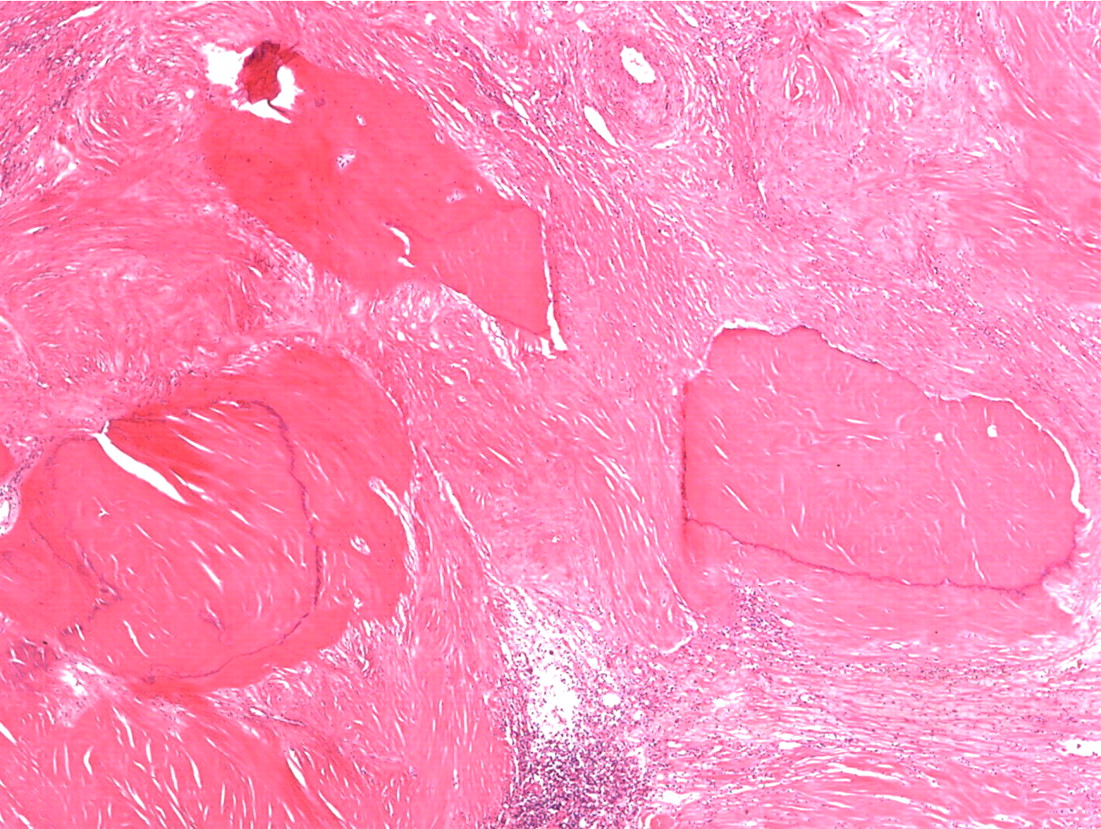
 Fig. 1-A
Fig. 1-A Fig. 1-B
Fig. 1-B Fig. 2
Fig. 2 Fig. 3-A
Fig. 3-A Fig. 3-B
Fig. 3-B Fig. 3-C
Fig. 3-C Fig. 3-D
Fig. 3-D Fig. 3-E
Fig. 3-E Fig. 3-A
Fig. 3-A Fig. 3-B
Fig. 3-B Fig. 3-C
Fig. 3-C Fig. 3-D
Fig. 3-D Fig. 3-E
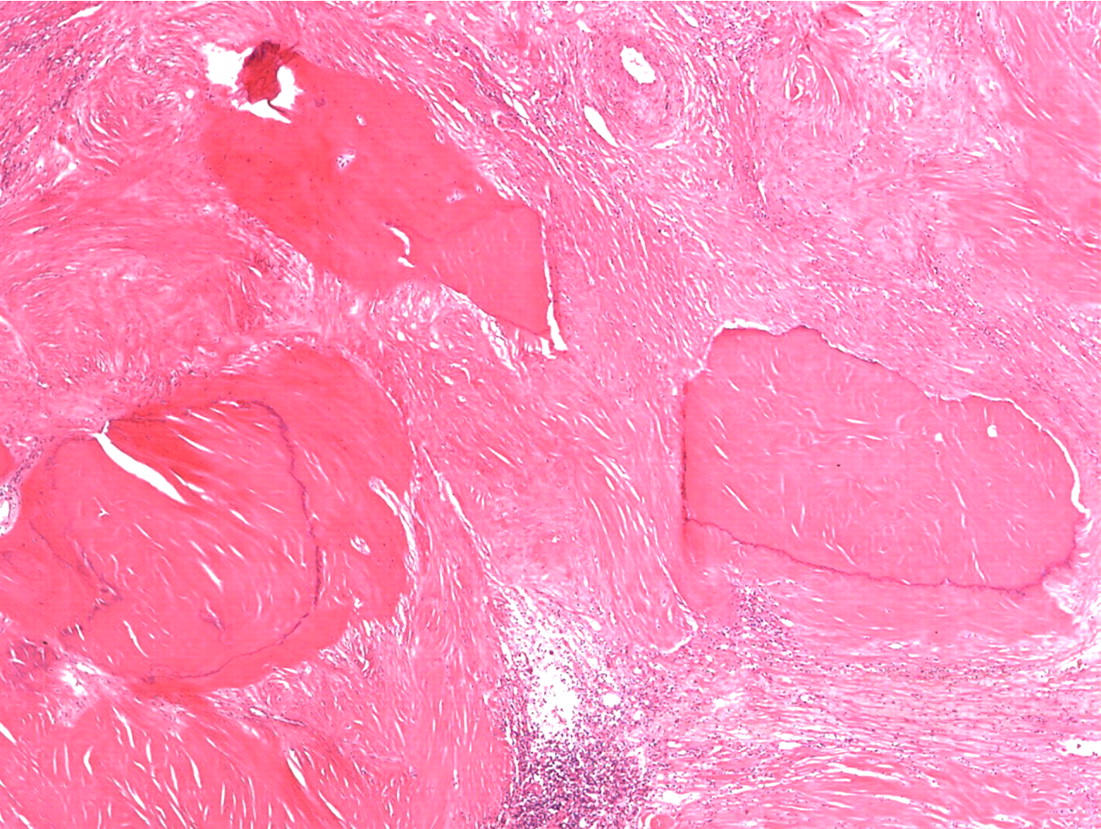
Fig. 3-EA sixty-three-year-old right-hand-dominant woman presented to our institution for evaluation and treatment of a complex deformity of the right upper extremity. Distinctive features of the deformity at the time of presentation included multiple firm nodular soft-tissue masses located on the dorsal and volar surfaces of the right hand and wrist, involving the thumb, index finger, and long fingers. The patient had severe contractures of the index finger and first web space in areas of particularly large and dense nodule formation; small skin ulcerations that appeared to be chronic and infected were seen in these areas and at the volar base of the first metacarpal (Figs. 1-A, 1-B, and 2). The patient had a concomitant dense sensory and motor neuropathy of the median nerve of the right hand. Full physical examination revealed other areas of hard, nodular swelling, specifically in the patient’s right shoulder, right upper arm, right lateral forefoot, and right iliac crest. Notably, the affected areas were limited to the right hemicorporeum. The medical history of the patient included an uneventful birth, no congenital skeletal malformations, and a normal early childhood development. The family history did not include hormone insensitivity, Albright hereditary osteodystrophy, or any disorders of extraskeletal ossification. At the time of presentation, the patient had three living children who did not display any signs of heterotopic ossification or any findings consistent with fibrodysplasia ossificans progressiva, progressive osseous heteroplasia, Proteus syndrome, or Albright hereditary osteodystrophy. At eight years of age, the patient had experienced a transient episode of weakness in the right arm that lasted for two weeks. The cause of this incident was not fully investigated at the time, and the symptoms resolved spontaneously after a series of chiropractic manipulations of the right upper extremity. When the patient was thirty years of age, a firm, exophytic lesion had developed on the right small toe. This lesion was excised, but multiple nodular mineralized masses developed at the surgical site. Despite re-excision, the masses recurred. When the patient was thirty-four years of age, pain and weakness had developed in the right upper extremity. Radiographs made at the time revealed a simple cyst in the proximal portion of the right humerus, and the patient subsequently underwent an incisional biopsy. Histologic evaluation of the biopsy tissue was consistent with the diagnosis of a simple bone cyst. Over the following two years, the cystic lesion in the proximal portion of the humerus persisted and additional cystic lesions developed in the right lateral epicondyle and the distal aspect of the radius. Both humeral lesions were treated with curettage and packing with use of autogenous cancellous bone graft obtained from the right anterior iliac crest. Firm, nodular, ossified soft-tissue masses developed postoperatively at all three surgical sites. When the patient was forty-three years of age, a new enlarging soft-tissue mass, measuring >10 cm in diameter, had been excised from the right arm. Shortly thereafter, another lesion developed on the right index finger; this mass was excised and the resultant skin defect was treated with a skin graft. Six years later, the right small toe was amputated because of an enlarging soft-tissue lesion; the surgical site was treated postoperatively with 2500 Gy of radiation. Medical records dating from the time of the right toe amputation documented that the lesion on the proximal part of the right humerus that had been excised six years before had recurred and was "the size of a grapefruit." Unfortunately, radiographs of this lesion are no longer available for review. "Atypical neurofibromatosis" had been the proposed diagnosis. One year later, the patient underwent a right carpal tunnel release. Sensory and motor dysfunction of the median nerve was documented with preoperative electromyography. Coincident with the carpal tunnel release, a 4 × 5-cm mass was excised from the palmar aspect of the index finger. Despite two postoperative doses of radiation (2500 Gy and 2100 Gy), the lesion of the index finger recurred and caused a flexion contracture of the proximal interphalangeal joint. At sixty-three years of age, the patient was referred to our institution for evaluation. Physical examination revealed numerous hard, nodular lesions of the right shoulder, elbow, hand, foot, and iliac crest. There were small draining skin ulcerations over prominent nodular lesions in the thenar area of the hand and on the lateral surface of the right foot. A technetium-99 radionuclide bone scan revealed increased uptake in all of the nodular areas. Laboratory evaluation of serum calcium, phosphorus, alkaline phosphatase, lactic dehydrogenase, C-reactive protein, and parathyroid hormone levels were normal. A twenty-four-hour urine collection revealed normal amounts of hydroxyproline, calcium, potassium, and phosphate. An electromyographic study and a nerve-conduction study revealed motor and sensory abnormalities of the right axillary, radial, musculocutaneous, and median nerves; these findings were consistent with multiple mononeuropathies rather than a brachial plexus lesion. A consultation with the Endocrinology Service resulted in the suggested diagnosis of atypical myositis ossificans. The patient was started on oral bisphosphonate therapy. The ossific lesions did not regress, however. Drainage persisted from the right thenar area, and the contracture at the proximal interphalangeal joint of the right index finger and the first web space worsened, resulting in a loss of functional pinch. Approximately one year after referral to our institution, the patient underwent excision of the right second ray and wide resection of the calcific masses over the dorsal and volar aspects of the right hand and wrist. Microneurolysis of the median nerve was also performed. The nerve was encased in mineralized tissue throughout its distal course. Because of the presence of thenar-muscle atrophy, local infection, and nodular mineralization, it was necessary to route the index profundus tendon around a pulley created with part of the flexor carpi ulnaris in order to restore thumb opposition. A capsulotomy of the carpometacarpal joint of the thumb was also performed in order to place the thumb in a more functional position. The resulting soft-tissue defect was covered with a latissimus dorsi muscle flap that was harvested from the unaffected left side of the patient, and vascular anastomoses were created in an end-to-side fashion to the ulnar artery in the mid-part of the forearm and to a large branch of the forearm venous system. A nonmeshed split-thickness skin graft, harvested from the contralateral buttock region, was applied for muscle coverage. Surgical pathology specimens consisting of excised tissue were submitted for examination (Figs. 3-A through 3-E).
Histopathologic analysis revealed multiple nodules of hyalinized fibrous tissue involving the skin, subcutaneous tissue, and tendons. The nodules were composed of hypocellular fascicles of fibroblasts that were interspersed between broad bundles of hyalinized collagen. In many of these nodules, a gradual or sharp transition to deposits of bone was present (Figs. 3-A through 3-E). In some areas, sheet-like deposits of woven bone were present, while in other areas within the same lesion, cortical and cancellous lamellar bone with interspersed fatty marrow was present. No cellular atypia or neural components were observed. There were no postoperative complications, and the patient recovered well on a maintenance dose of etidronate (400 mg orally, twice daily for six months following surgery). At the time of the five-year follow-up, there was no evidence of recurrence at the operative site, although a small (0.5 × 1.0 cm) nodule had developed just adjacent to the flap on the dorsum of the hand. Hand function had improved considerably, as the surgery had restored side-to-side pinch. The patient was using the hand mostly in the performance of assistive functions. She had recovered only protective sensation in the thumb and long finger. Similarities between the syndrome that affected our patient and progressive osseous heteroplasia prompted us to pursue a genetic diagnosis. The recent identification of mutations in GNAS1, the gene that encodes the alpha subunit of the stimulatory G protein of adenylyl cyclase, as the cause of progressive osseous heteroplasia has enabled molecular screening for progressive osseous heteroplasia. Samples of genomic DNA isolated from peripheral blood samples taken from our patient were screened by one of the authors (E.M.S.) for mutations in the GNAS1 gene. No mutations were identified in the coding sequence of the GNAS1 gene in our patient. The diagnosis of a new syndrome entitled focal fibronodular heterotopic ossification was made.
Proceed to Discussion >>Reference: Yeon HB, Kaplan FS, Shore EM, Rosenberg AE, Jupiter JB. Focal fibronodular heterotopic ossification. A case report. J Bone Joint Surg Am. 2007 Jun;89(6):1329-36.
In this case report, we present a new syndrome of heterotopic ossification that involved the right side of our patient. This new condition, which we have termed "focal fibronodular heterotopic ossification," shares some clinical findings with previously described disorders of heterotopic ossification, including intra-articular and extra-articular chondromas and osteochondromas, extraskeletal chondromas, osteomas, myositis ossificans, pseudomalignant heterotopic ossification, and extraosseous osteosarcoma. It is also similar to rare genetic syndromes, including fibrodysplasia ossificans progressiva, progressive osseous heteroplasia, juvenile hyaline fibromatosis, and Proteus syndrome, although it is clearly a distinct entity. Fibrodysplasia ossificans progressiva is a rare genetic disorder that was first described by Guy Patin in 1648 and that is characterized by progressive extraskeletal ossification heralded by congenital malformation of the great toes, thumbs, hips, and cervical spine. Fibrodysplasia ossificans progressiva may arise spontaneously or may be inherited in an autosomal dominant manner with variable expression and complete penetrance. The classic diagnostic triad for this disease consists of radiographic abnormalities of the hands or feet; progressive heterotopic endochondral ossification of skeletal muscle, tendon, and fascia; and a predictable pattern of involvement that progresses from axial to appendicular, proximal to distal, and cranial to caudal. Increased heterotopic bone formation is observed in response to local trauma, and extraskeletal ossification can lead to severe contractures. Patients with fibrodysplasia ossificans progressiva have bilateral involvement of muscle, tendons, and ligaments and a classically endochondral pattern of ossification. Despite some similarity between fibrodysplasia ossificans progressiva and focal fibronodular heterotopic ossification, the diagnosis of fibrodysplasia ossificans progressiva could be excluded in our patient on the basis of a lack of congenital malformations of the toes, a lack of involvement of the axial skeleton and chest wall, a lack of bilateral involvement, and a predominantly intramembranous pattern of ossification. Two disease loci have been genetically mapped in kindred with fibrodysplasia ossificans progressiva, and a disease-causing mutation for fibrodysplasia ossificans progressiva has recently been identified in the glycine-serine activation domain of ACVR1, the gene that encodes the activin A type-I receptor. ACVR1 is a receptor for bone morphogenetic protein (BMP), which supports the hypothesis that fibrodysplasia ossificans progressiva results from misregulated BMP signaling. Recently, Ahn et al. demonstrated that the negative feedback antagonist response to bone morphogenic protein-4 (BMP-4) stimulation is dramatically reduced in fibrodysplasia ossificans progressiva cells compared with control cells. Kan et al. further reported that progressive extraskeletal ossification develops in transgenic mice that overexpress BMP-4 under control of the neuron-specific enolase promoter, albeit in a pattern distinctly different from that observed in animals with fibrodysplasia ossificans progressiva. De la Pena et al. reported profound abnormalities in BMP-receptor trafficking and downstream signaling in fibrodysplasia ossificans progressiva cells. The pattern of expression of the neuron-specific enolase promoter, principally in neurons and neuroendocrine cells, suggests that peripheral nerves may play a role in the pathogenesis of heterotopic ossification. This hypothesis is further bolstered by the long-standing observation that specific disorders of the nervous system, such as closed head trauma and spinal cord injuries, can lead to heterotopic ossification. Progressive osseous heteroplasia, first described as a syndrome in 1994 by Kaplan et al., is a rare genetic disorder characterized by progressive heterotopic ossification involving skin, subcutaneous fat, and deep connective tissue. The onset of progressive osseous heteroplasia is usually during infancy. Maculopapular rashes presage islands of heterotopic bone that later coalesce to form large ossified plaques that slowly but inexorably progress to involve deep connective tissues, leading to ankylosis of adjacent joints and retardation of limb growth. Progressive osseous heteroplasia is distinguishable from fibrodysplasia ossificans progressiva by bone deposition in the skin, the absence of congenital skeletal malformations, the lack of preosseous tumorlike swelling, and the presence of primarily intramembranous rather than endochondral ossification. In light of these findings, progressive osseous heteroplasia is clinically more similar to focal fibronodular heterotopic ossification than is fibrodysplasia ossificans progressiva, although progressive osseous heteroplasia can clearly be distinguished from the condition seen in our patient on the basis of clinical criteria. For example, the onset of symptoms in our patient was at thirty years of age rather than in infancy, and triggering factors, such as trauma or surgery, which do not characteristically lead to additional progressive osseous heteroplasia lesions, led to additional ossified nodules in adjacent tissues in our patient. In addition, large nodular lesions of heterotopic bone are not observed in progressive osseous heteroplasia. Juvenile hyaline fibromatosis is a rare inherited connective-tissue disorder with onset during infancy or early childhood. It was first described by Murray in 1873 under the name “molluscum fibrosum,” but it was later called "Puretic syndrome." Characteristic clinical findings in patients with juvenile hyaline fibromatosis include pearly skin papules or subcutaneous nodules; plaques on the back, ears, scalp, and hands; joint contractures; and gingival hypertrophy. Histologically, the lesions show an abundant eosinophilic matrix with embedded spindle and epithelioid cells without nuclear atypia. Multinucleated giant cells with intracytoplasmic granules are also present. Extraskeletal ossification has not been associated with juvenile hyaline fibromatosis lesions. In five families with juvenile hyaline fibromatosis, mutations in the capillary morphogenesis factor-2 gene (CMG2) have been identified, and dysregulation of basement membrane architecture resulting from these mutations has been proposed as an important etiologic factor. Our patient did not exhibit clinical findings consistent with juvenile hyaline fibromatosis (i.e., early onset, skin papules, and gingival hypertrophy); and her condition was further distinguishable by the prominent presence of ossification. Proteus syndrome, first described by Cohen and Hayden in 1979, is a hamartomatous disorder characterized by connective tissue nevi, epidermal nevi, hemihypertrophy, hyperostoses, lipomas, and vascular malformations that are present at birth and progress thereafter. Named by Wiedemann in 1983 after a Greek god who could change his appearance at will, Proteus syndrome has historically posed a difficult diagnostic challenge due to the broad spectrum of clinical presentations that it encompasses. More recently, Proteus syndrome has been defined more rigorously with use of a combination of general and specific criteria. General or mandatory criteria for the diagnosis of Proteus syndrome are a mosaic distribution of lesions, sporadic occurrence, and progressive worsening—all of which were met in the case of our patient. Specific criteria include cerebriform connective-tissue nevi, epidermal nevi, hemihypertrophy, skull hyperostosis, megaspondylodysplasia, splenomegaly, vascular malformation, dysmorphic facies, and characteristic tumors including ovarian cystadenoma and parotid monomorphic adenoma. Our patient did not display any of these specific criteria. Mutations in the tumor suppressor gene PTEN have been reported in some patients with Proteus syndrome. Because mutations in this gene have not been found in all patients with clinically diagnosed Proteus syndrome, it is possible that the disorder may be caused by mutations in more than one gene or that the current clinical diagnostic criteria may be overly inclusive. Although the genes responsible for rare syndromes of heterotopic ossification have not been fully identified, recent progress in this area makes a genetic etiology for dysregulated extraskeletal ossification seem likely. In our patient, the sporadic occurrence and strict confinement of symptoms to the right hemicorporeum is suggestive of an etiology involving somatic mosaicism, as in the McCune-Albright syndrome in which activating mutations of GNAS1 found only in affected tissues cause polyostotic fibrous dysplasia, café au lait skin patches, and endocrine abnormalities, while germline mutations are not observed and are believed to be lethal early in embryogenesis. Asymmetric expression itself, however, does not prove genetic mosaicism, as patients with progressive osseous heteroplasia with germline inactivating mutations in GNAS1 show dramatic differences in phenotype from side to side. Notably, a patient with strictly hemimelic progressive osseous heteroplasia has been described; however, tests for genetic mosaicism were not reported. Other musculoskeletal disorders, including Klippel-Trénaunay-Weber syndrome, neurofibromatosis, localized giantism, and hemihypertrophy, may also exclusively affect one side of the body, although the molecular causes of these conditions are not fully characterized. Asymmetry may suggest that the disease-causing mutation is necessary but not sufficient for expression of the disease phenotype. Existing literature regarding progressive heterotopic ossification documents few effective medical or surgical treatments. Treatment of fibrodysplasia ossificans progressiva with oral bisphosphonates has generally been associated with poor results. We undertook operative intervention in the case of our patient to eliminate a chronic infection and simultaneously to improve the function and appearance of what had become a disfigured and disabled hand. A contralateral latissimus dorsi muscle flap was chosen, among other reasons, to avoid use of tissues that might be affected by a disease-causing somatic mutation. Eventually, more effective treatments will be based on a fundamental understanding of the molecular etiology of this and other related conditions of progressive heterotopic ossification.
Reference: Yeon HB, Kaplan FS, Shore EM, Rosenberg AE, Jupiter JB. Focal fibronodular heterotopic ossification. A case report. J Bone Joint Surg Am. 2007 Jun;89(6):1329-36.



Fibrodysplasia ossificans progressive
Proteus syndrome
Intra-articular and extra-articular chondromas and osteochondromas
Progressive osseous heteroplasia
Focal fibronodular heterotopic ossification