An Eight-Year-Old Girl with Progressive Hip Pain
December 17, 2014
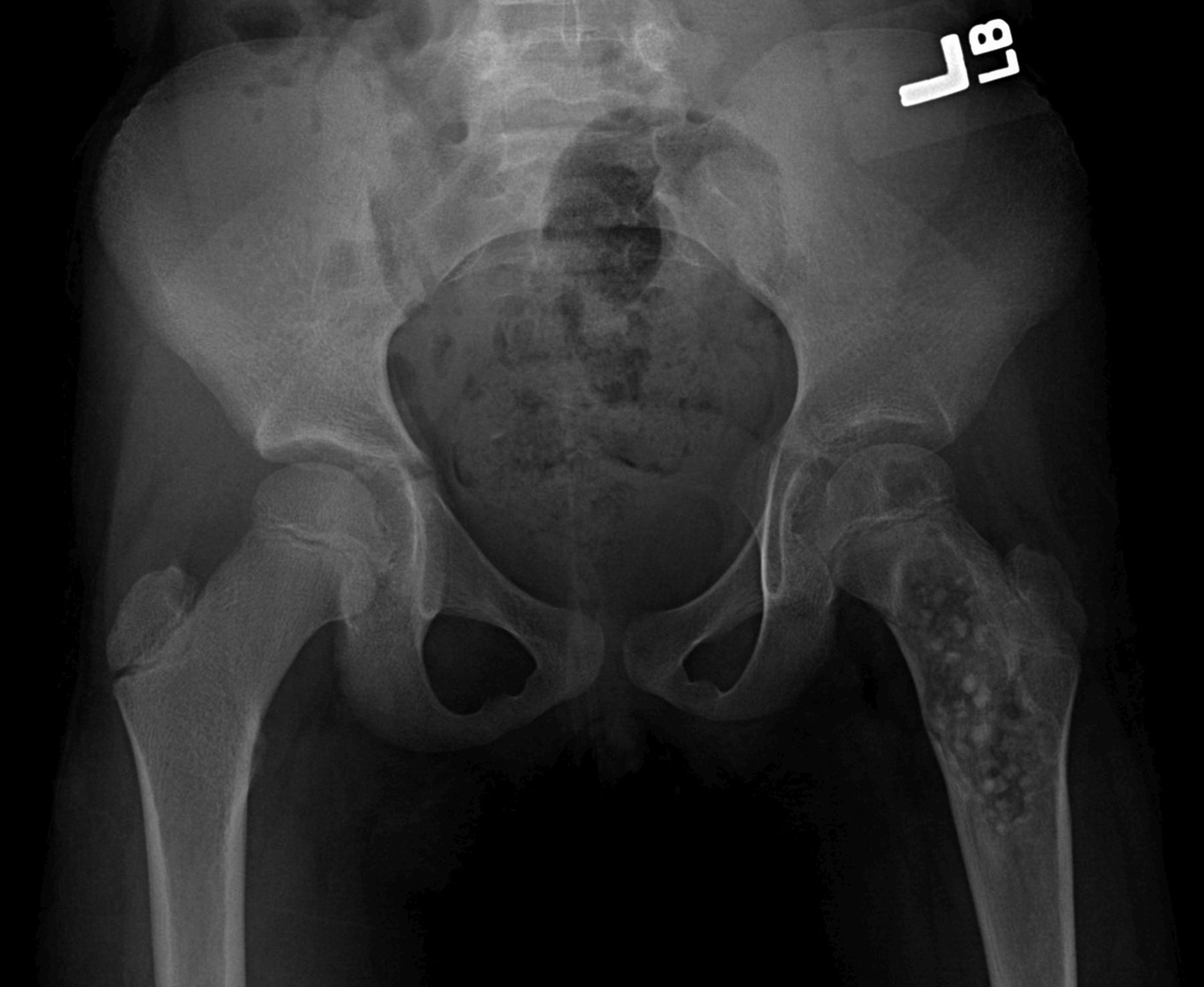
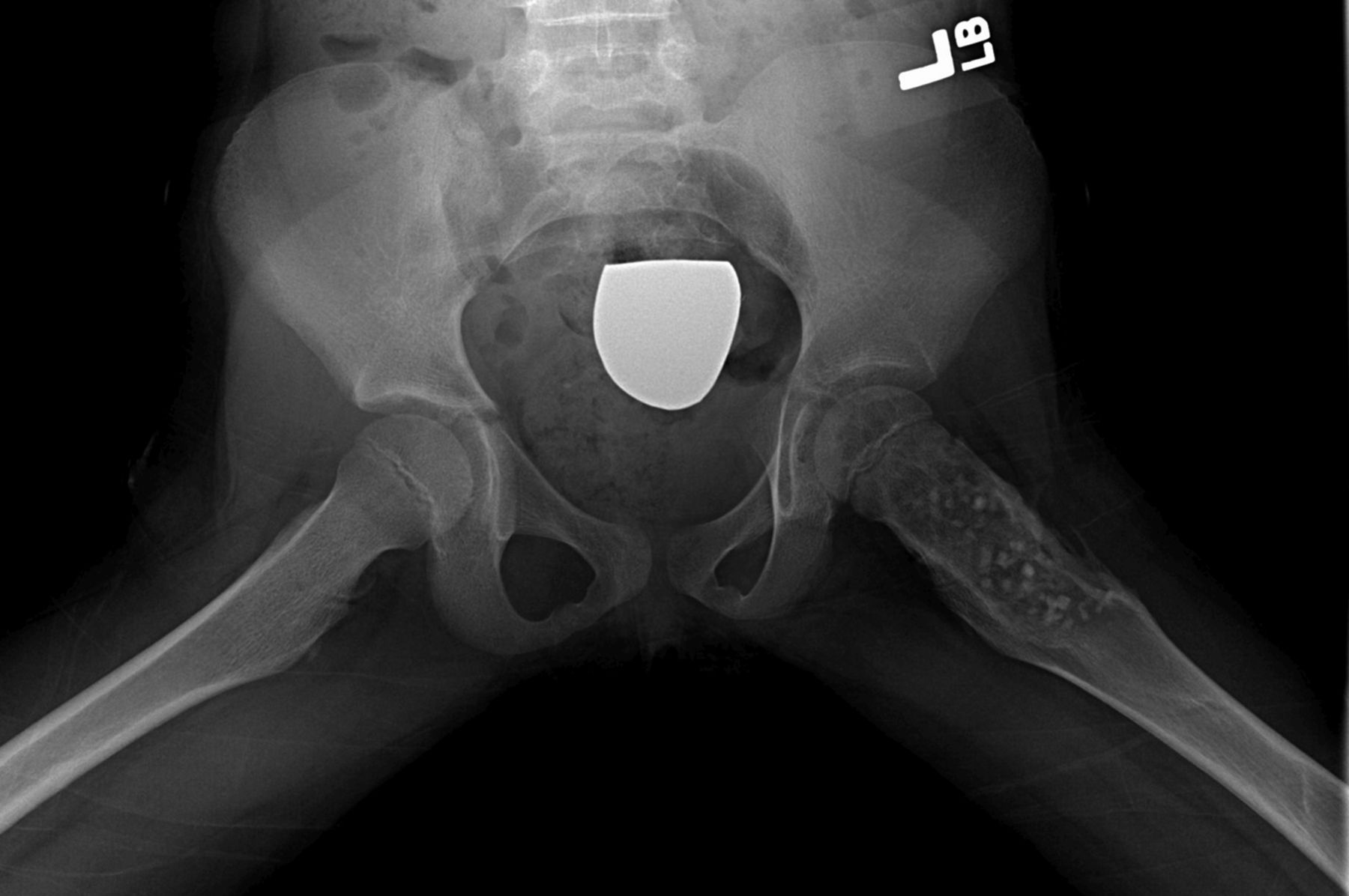
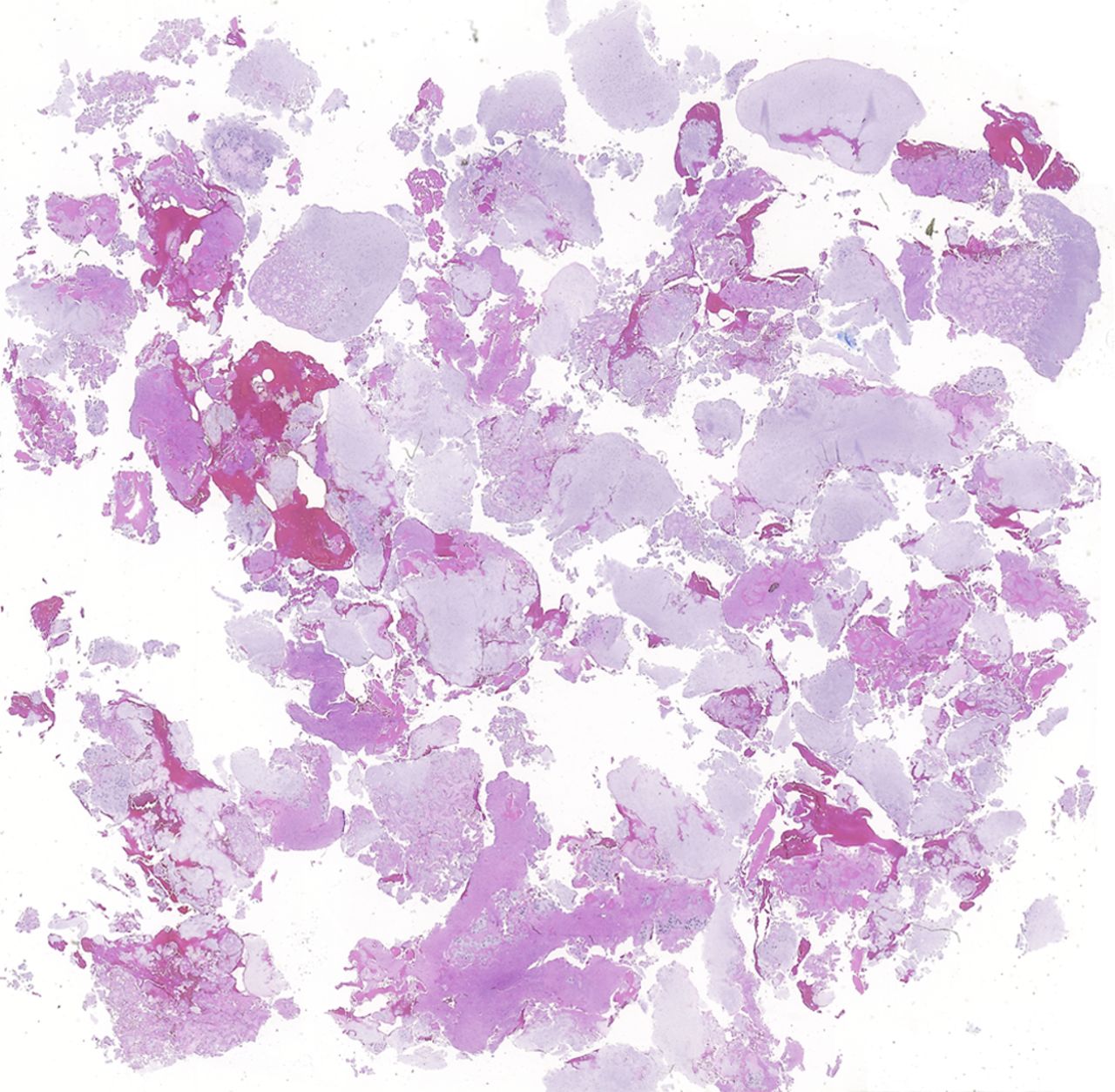
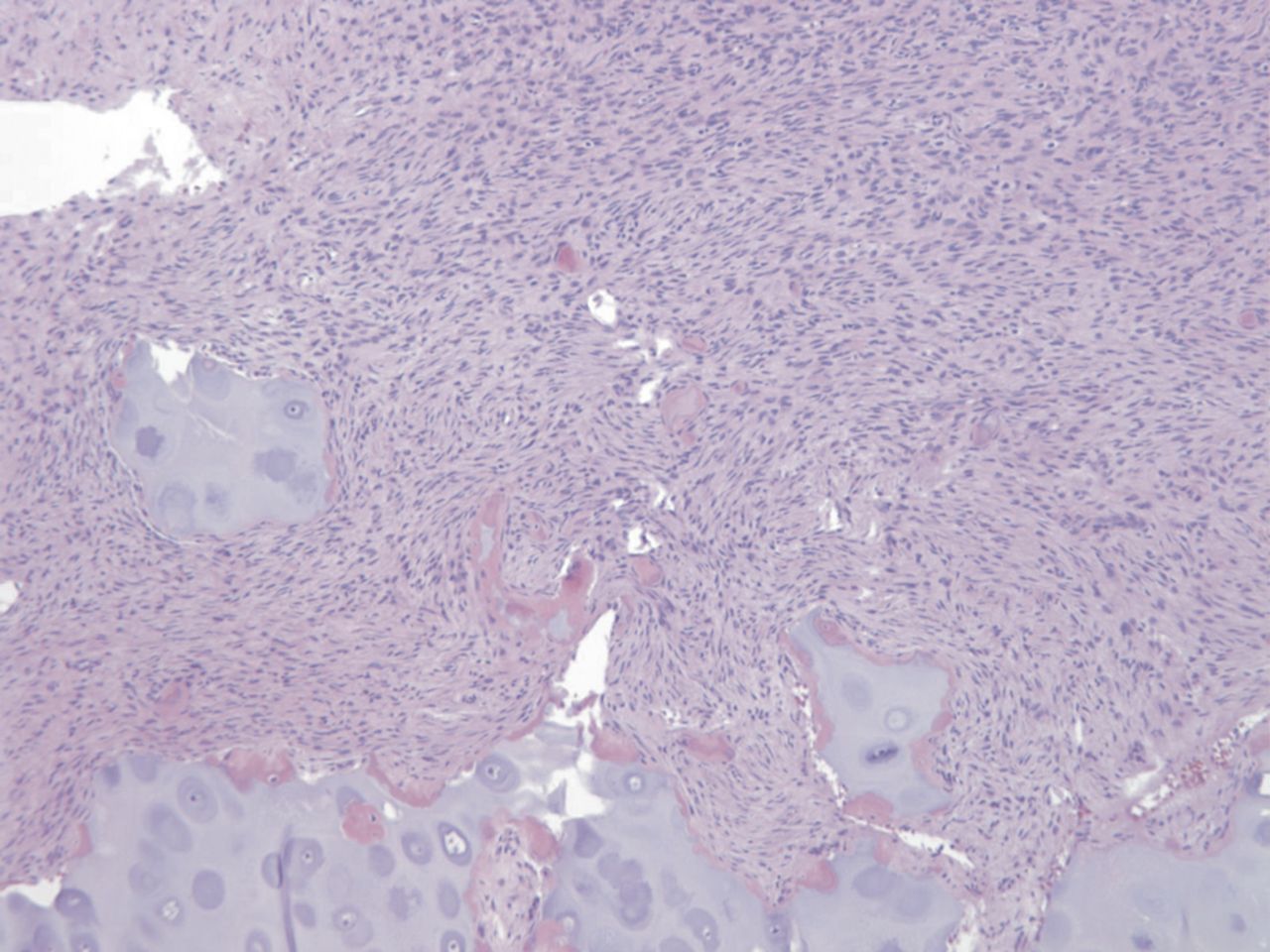
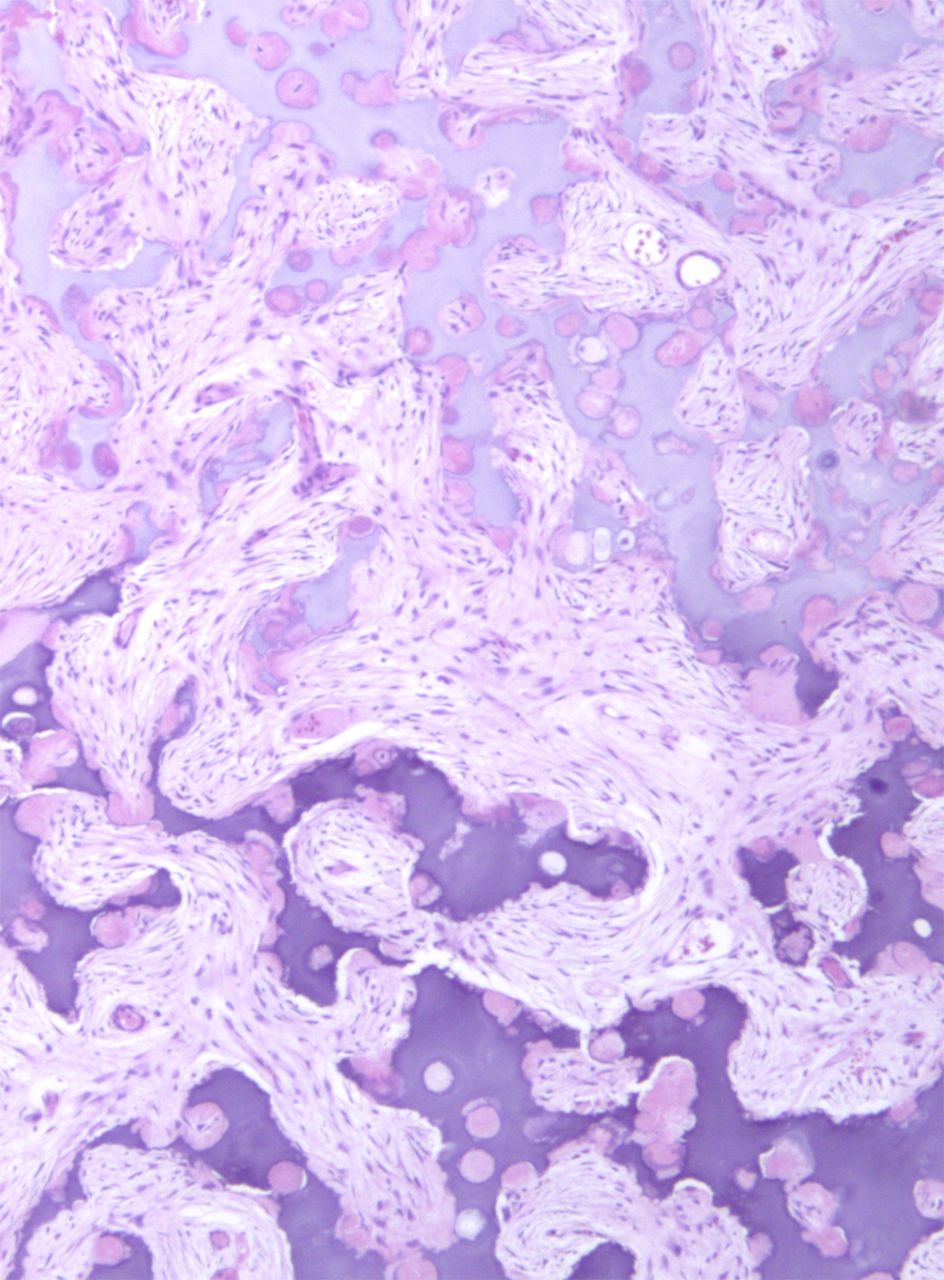
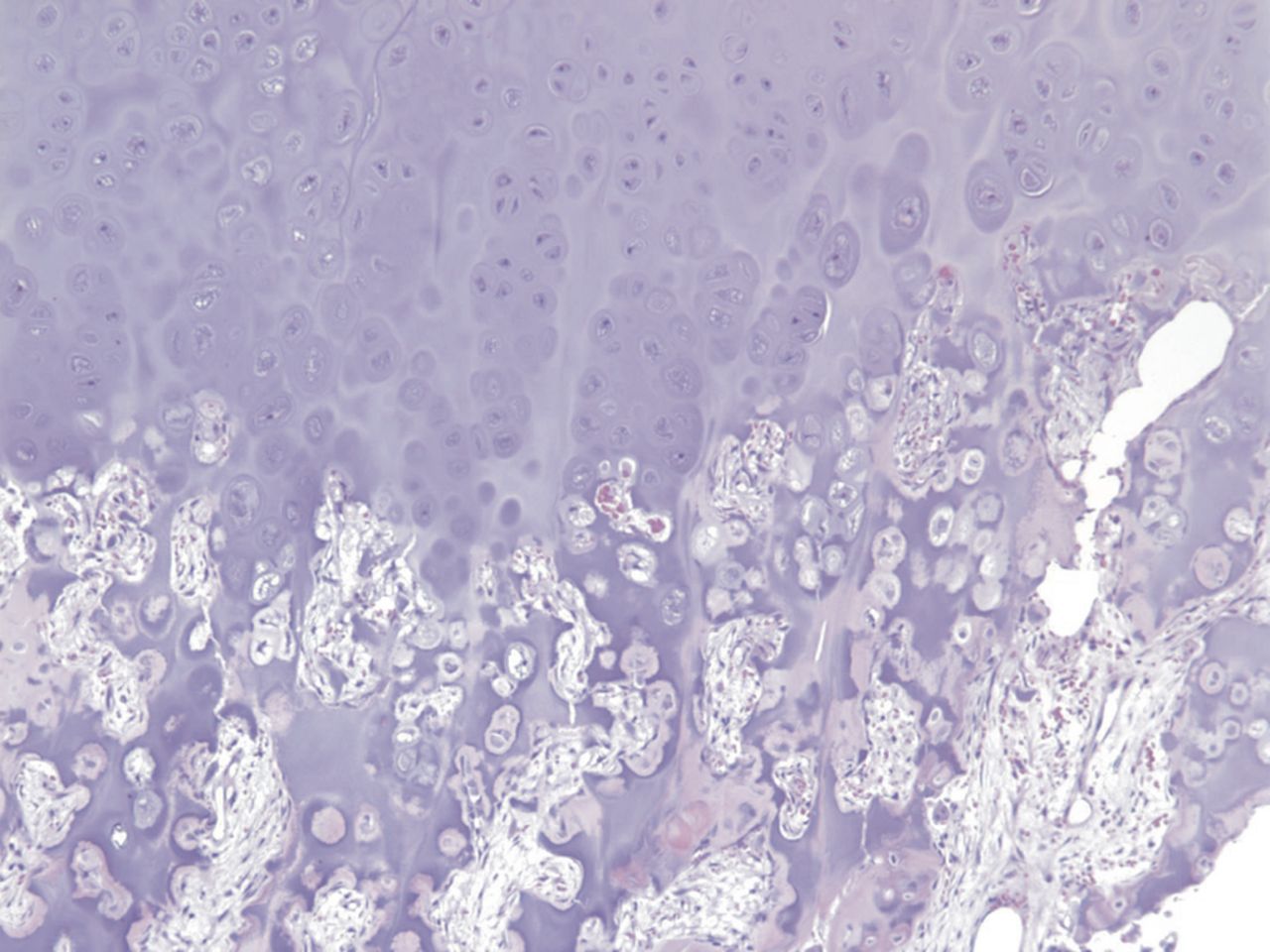
 Fig. 1-A
Fig. 1-A Fig. 1-B
Fig. 1-B Fig. 2-A
Fig. 2-A Fig. 2-B
Fig. 2-B Fig. 2-C
Fig. 2-C Fig. 2-D
Fig. 2-D Fig. 2-A
Fig. 2-A Fig. 2-B
Fig. 2-B Fig. 2-C
Fig. 2-C Fig. 2-D
Fig. 2-D Fig. 3-A
Fig. 3-A Fig. 3-B
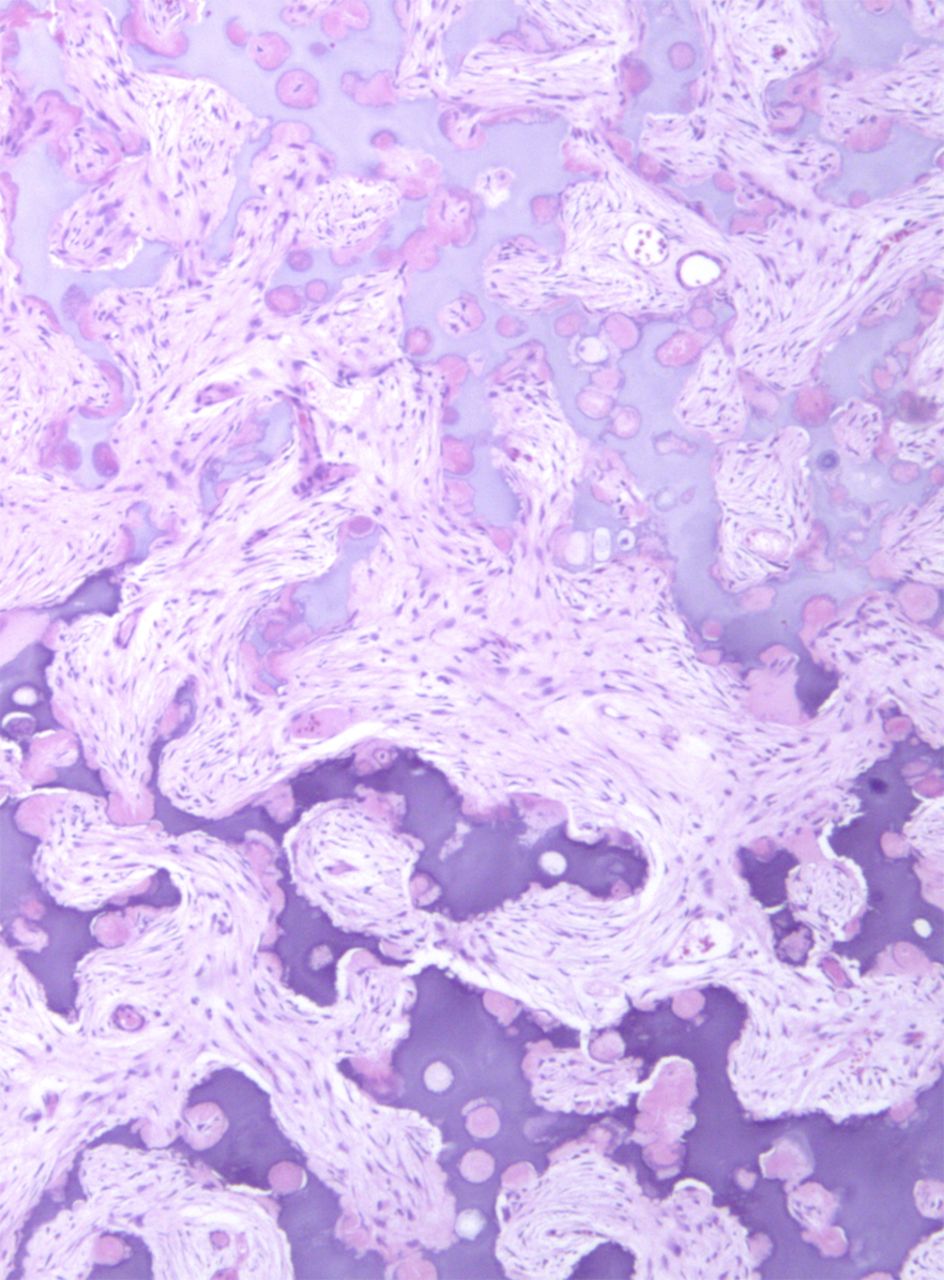
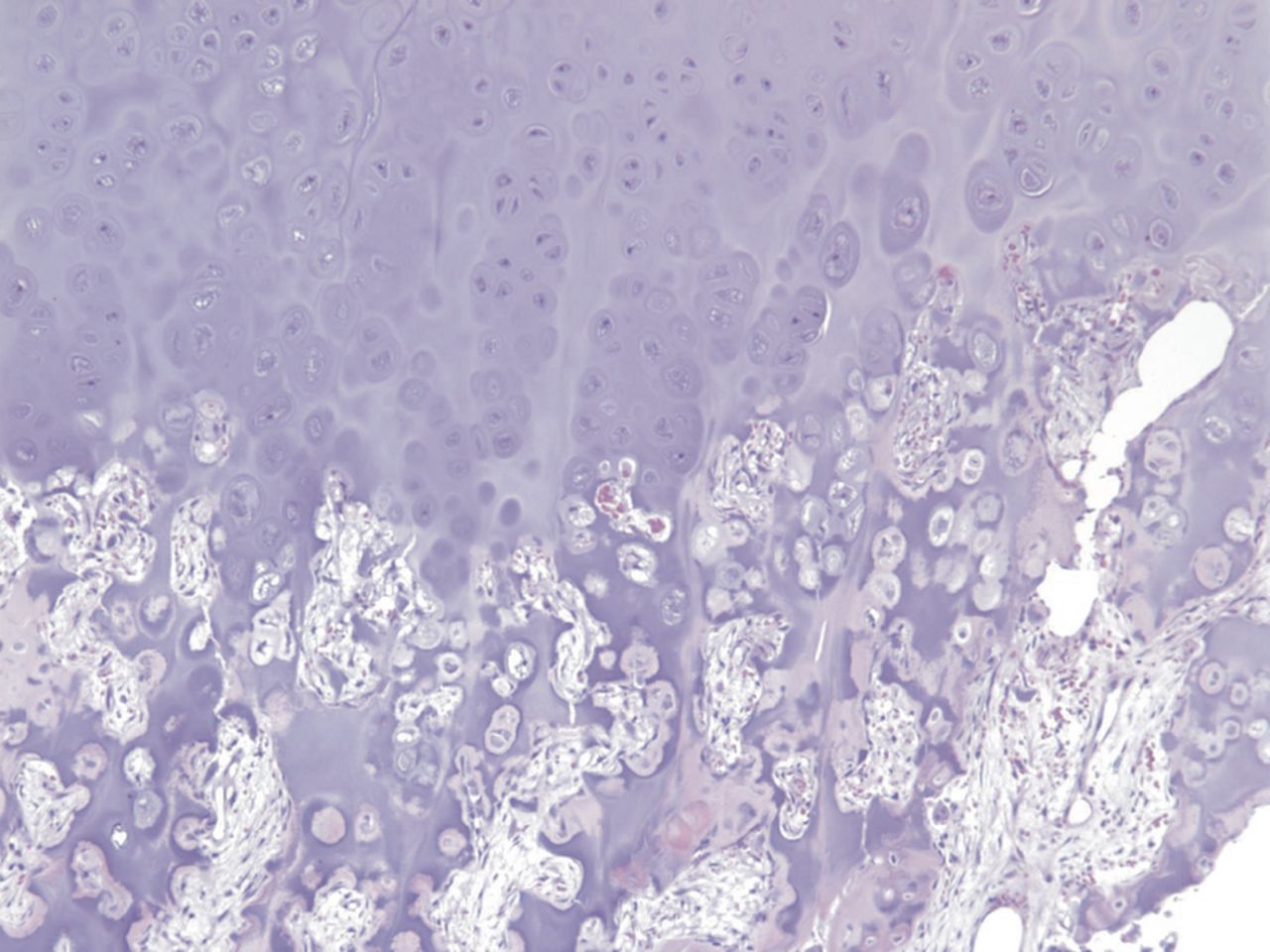
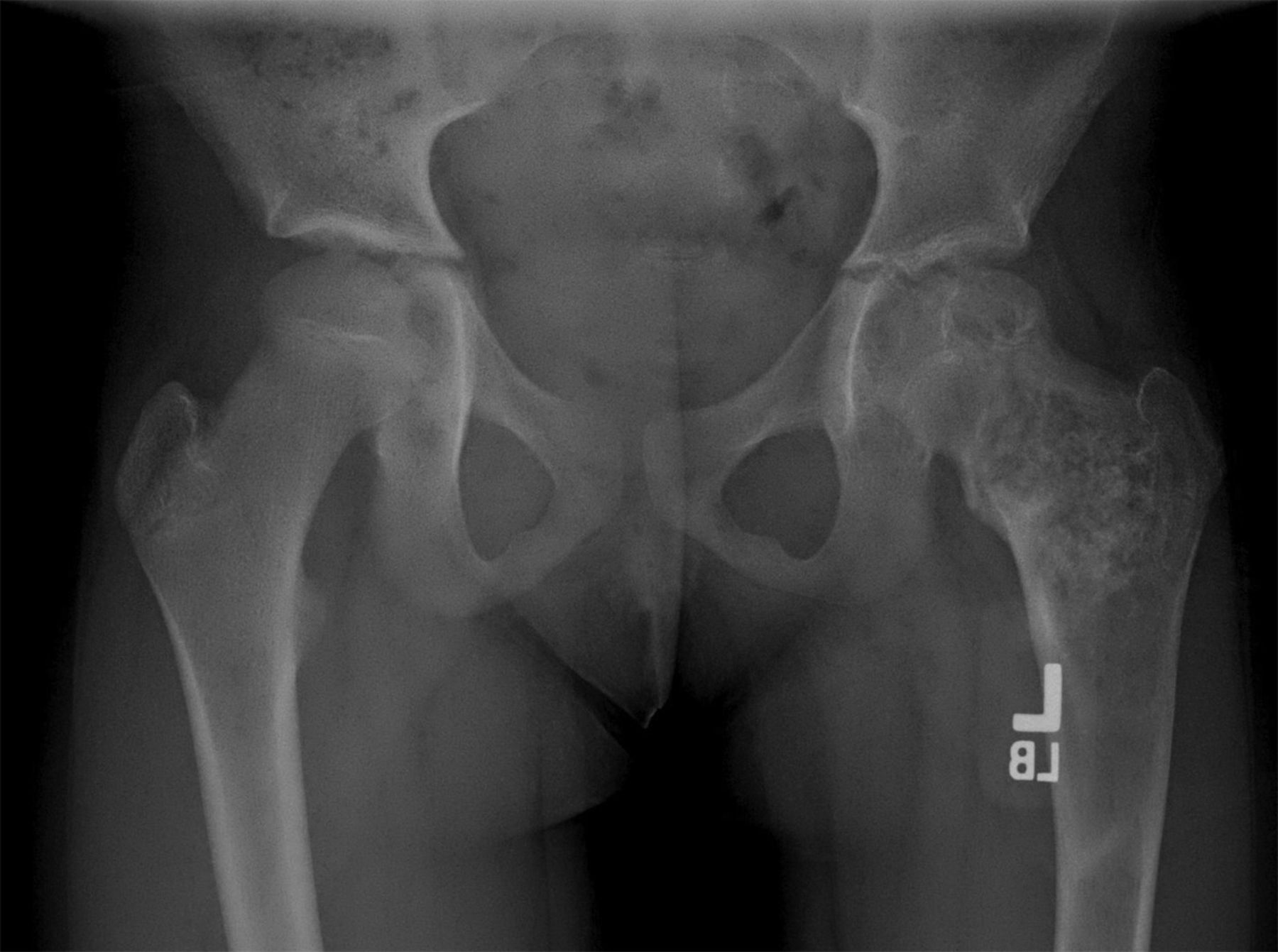
Fig. 3-BAn eight-year-old girl presented with an eight-week history of a limp and intermittent but progressive left hip pain associated with activity but not at rest. She was administered over-the-counter analgesic medication without relief. She denied antecedent trauma, constitutional symptoms, or recent illness. She localized the pain to the left groin and greater trochanter. The past medical history was unremarkable. During physical examination, the patient’s gait was antalgic, and she allowed full active and passive range of motion with discomfort at the extremes of internal and external rotation, flexion, and extension. The neurovascular examination was normal. The skin overlying the hip did not demonstrate abnormalities; no mass, tenderness, muscle atrophy, or other external abnormality was detected. Family and past medical history were unremarkable. Anteroposterior and frog-leg lateral radiographs of the pelvis demonstrated a 7 cm × 2-cm mixed radiolucent and mineralized lesion with stippled calcifications involving the left femoral neck and peritrochanteric region; a radiolucent region was seen within the femoral epiphysis (Figs. 1-A and 1-B). A fracture involving the medial femoral neck was present. The lesion demonstrated a variable transition zone defined by an intermittent sclerotic rim, no appreciable periosteal reaction, no soft-tissue mass, and moderate cortical scalloping. Extralesional portions of the femur showed cortical irregularity. Based on the radiographic findings, the lesion was presumed to be benign; open biopsy was recommended. After informed consent and preoperative evaluation, the patient was brought to the operating room where a biopsy was undertaken through a lateral approach to the femur. In the event of a malignant diagnosis, the lateral approach would have allowed a limb-sparing proximal femoral replacement performed through an extensile lateral or posterolateral incision. The histologic specimens are shown in Figs. 2-A through 2-D.
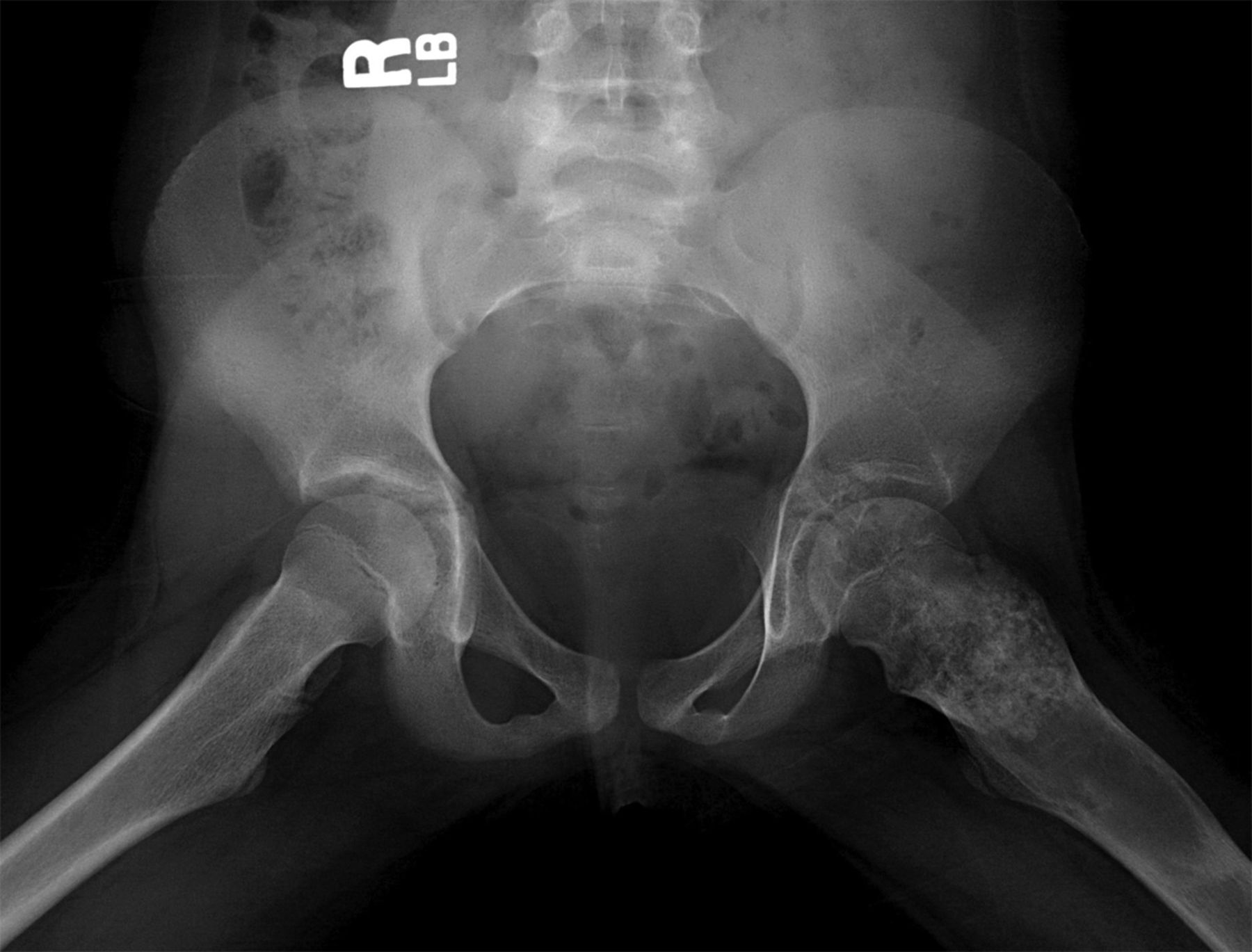
Permanent section analysis revealed a fibro-osseous lesion with a massive cartilaginous component as the dominant element (Fig. 2-A). In some areas, the appearance was that of conventional fibrous dysplasia and fibrocartilaginous dysplasia. Other areas, however, showed stromal hypercellularity in a monotonous pattern reminiscent of fibrocartilaginous mesenchymoma (Fig. 2-B). Heterogeneous areas of intervening collagen, lamellar and woven bone, hemosiderin, and endochondral ossification were seen (Fig. 2-C); no conspicuous areas of mitotic activity or anaplasia were noted. Rare osseous constituents were predominantly contiguous woven and lamellar bone rimmed by occasional osteoblasts. The chondroid matrix constituted the majority of the specimen and consisted of disorganized hyaline cartilage that, in some areas, was reminiscent of epiphyseal plates and endochondral ossification; no atypical features were noted (Fig. 2-D). Following treatment, a tumor specimen embedded in optimal cutting temperature compound was submitted for GNAS mutation analysis. DNA was extracted from approximately 100 mg of tissue with use of proteinase K lysis with the iPrep ChargeSwitch Buccal Cell DNA extraction kit (Invitrogen, Carlsbad, California). The GNAS gene was analyzed with bidirectional Sanger sequencing (AB BigDye Chemistry, XTerminator Purification Kit) on a 3500xL automated genetic analyzer (Applied Biosystems, Foster City, California), according to the manufacturer’s protocols. Sequences were analyzed with use of reference files NG_016194.1 (genomic) or NM_000516.4 messenger RNA (mRNA) on Mutation Surveyor software (SoftGenetics, State College, Pennsylvania). DNA analysis revealed a mutation at codon 201 at exon 8, substituting cysteine for arginine. This mutation is frequently identified in fibrous dysplasia. We treated our patient with intralesional curettage. After irrigating, we filled the lesion with allograft croutons and demineralized bone matrix (BioHorizons, Toronto, Ontario, Canada); no adjuvant was used. The postoperative course was uneventful. She continued toe-touch weight-bearing with use of a walker for a total of eight weeks, at which time she was advanced to full weight-bearing as tolerated and the use of a walker. Three months after surgery, she had full hip motion, no tenderness, and could bear weight without restrictions. At the three-year follow-up, she reported no hip pain. On physical examination, the hip motion showed extension of 20° on both the operated and contralateral sides, flexion of 120° on both sides, internal rotation of 45° on the operated side and 75° on the contralateral side, and external rotation of 80° on the operated side and 90° on the contralateral side. Based on measurements from full-length lower-extremity radiographs, the patient had a 6-mm limb-length discrepancy (the left side was shorter than the right side). Anteroposterior (Fig. 3-A) and frog-leg lateral (Fig. 3-B) radiographs three years after surgery revealed good bone graft incorporation and no evidence of recurrence.
Proceed to Discussion >>Reference: Henderson ER, Monforte H, Neustadt JB. Fibrocartilaginous dysplasia of the proximal part of the femur with a pathological fracture in a child: A case report. JBJS Case Connect. 2014 Oct 08;4(4):e90.
Fibrous dysplasia and fibrocartilaginous dysplasia lesions are usually asymptomatic unless structural weakening causes mechanical pain; swelling may be apparent with expansile lesions, and fracture is rare. The onset of symptoms is generally insidious with progressive local pain and swelling, indicating a slow-growing lesion. The presentation of fibrocartilaginous mesenchymoma is similarly nonspecific. Our patient initially experienced unremarkable symptoms but progressed to a pathological fracture. Radiographs demonstrated a predominantly osteolytic metaphyseal lesion with focal mineralization, which characterizes this lesion. Intraoperative biopsy revealed a lesion with predominantly chondroid matrix, which was admixed with trabecular bone, and a bland-to-hypercellular fibrous stroma showing sparse polymorphism without mitotic figures, all of which are characteristic of this lesion; the diagnosis was supported by later confirmation of the GNAS mutation by polymerase chain reaction. Described treatments for fibrocartilaginous dysplasia include observation, curettage with bone-grafting, intramedullary nailing, and en bloc excision. Because of the overall benign radiographic appearance of our patient’s lesion, she was managed with open biopsy, frozen specimen analysis, and curettage. It should be emphasized that this method requires the close participation of an experienced musculoskeletal pathologist. In the absence of experienced musculoskeletal radiology and pathology teams, our patient could have been managed equally with a closed image-guided biopsy, followed by permanent specimen analysis and delayed definitive treatment. On imaging, this lesion demonstrated a transition zone defined by an irregular sclerotic rim without obvious periosteal reaction, absence of a soft-tissue mass, and intermittent cortical scalloping, clinically suggesting a histology of medium aggressiveness. Chondromyxoid fibroma is uncommon in the proximal part of the femur and generally has a lobulated appearance, a more homogeneous matrix, fibromyxoid stroma, and peripheral condensation of small spindle cells. Similar clinical, radiographic, and histopathologic features complicate distinguishing fibrocartilaginous dysplasia from fibrocartilaginous mesenchymoma. Fibrous dysplasia has a variable morphologic appearance, typically described as Chinese-character-shaped plates of woven bone without rimming osteoblasts amid relatively paucicellular fibrocollagenous stroma. On morphological grounds, the histologic differential diagnosis of conventional fibrous dysplasia includes low-grade intramedullary osteosarcoma. Depending on location, other conditions such as osteofibrous dysplasia, ossifying or cementifying fibroma, and other fibro-osseous lesions are also considered. Additional chondroid elements of fibrocartilaginous dysplasia may be subtle or, as seen in our patient, dominant, including epiphyseal plate morphology. While a large chondroid component is required for diagnosis of fibrocartilaginous dysplasia, no absolute percentage is defined, although 60% has been suggested. Our patient’s other potential diagnosis, fibrocartilaginous mesenchymoma, is a benign locally aggressive bone tumor of children and adolescents; to the best of our knowledge, only twenty cases have been reported in the literature. It was first described in 1984 when Dahlin et al. reported five cases of a previously unrecognized bone neoplasm that was called fibrocartilaginous mesenchymoma with low-grade malignancy. Bulychova et al. renamed the neoplasm fibrocartilaginous mesenchymoma after determining that the lesion has no potential for metastasis; the authors reported that local recurrence is possible. Fibrocartilaginous mesenchymoma has been described in the proximal part of the tibia, the proximal part of the humerus, the proximal part of the fibula, the fibular shaft, the distal part of the femur, the metatarsal bone, the ribs, the pubic bone, the iliac spine, the pubis, the thoracic vertebrae, and the lumbar vertebrae. Radiographs of fibrocartilaginous mesenchymoma classically demonstrate a poorly circumscribed, predominantly osteolytic metaphyseal lesion with coarsened trabeculae and focal mineralization. Computed tomography usually reveals irregular endosteal scalloping, and magnetic resonance imaging will demonstrate heterogeneous signal and can assist definition of marrow involvement and soft-tissue extension. Histologically, fibrocartilaginous mesenchymoma is characterized by three elements: hypercellular spindle cell proliferation with fibrosarcomatous morphology, trabecular bone production, and hamartomatous cartilage islands. Spindle cells are long, slender, and arranged in intersecting fascicles with sparse nuclear polymorphism and hyperchromatism. The cartilaginous islands are well-circumscribed nodules interspersed within the proliferating spindle cells or epiphyseal plates. Because of substantial overlap, it has been postulated that fibrocartilaginous dysplasia and fibrocartilaginous mesenchymoma are not separate entities. However, some pathologists suggest that the fibrosarcomatous morphology focally present in fibrocartilaginous mesenchymoma allows distinction from fibrocartilaginous dysplasia. Fibrocartilaginous mesenchymoma usually lacks deformity, may extend into soft tissue, and has no known polyostotic manifestation. To our knowledge, there are no reports of GNAS mutation analysis in cases of fibrocartilaginous mesenchymoma. GNAS mutation analysis is now being incorporated for the diagnosis of fibro-osseous craniofacial and long-bone lesions with considerable sensitivity and specificity. Of interest is a recent report of a similar GNAS mutation in parosteal osteosarcoma, reminding us that molecular diagnostics are to be used judiciously and in correlation with clinicopathologic parameters.
Reference: Henderson ER, Monforte H, Neustadt JB. Fibrocartilaginous dysplasia of the proximal part of the femur with a pathological fracture in a child: A case report. JBJS Case Connect. 2014 Oct 08;4(4):e90.
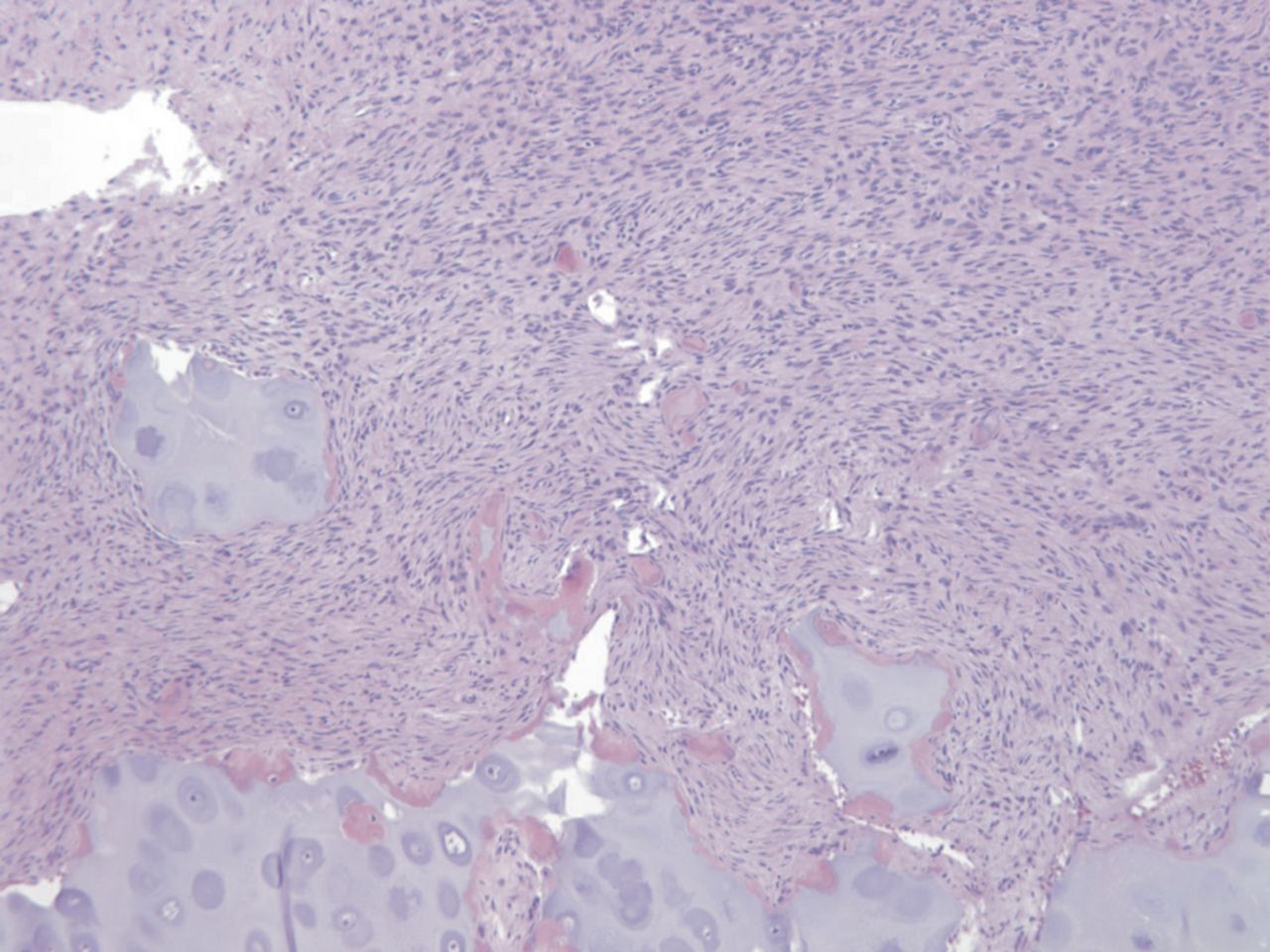
Chondroblastoma
Chondrosarcoma
Aneurysmal bone cyst
Simple bone cyst
Fibrocartilaginous dysplasia