A Neonate with a Parenchymal Mediastinal Mass
October 21, 2015

 Fig. 1
Fig. 1 Fig. 2
Fig. 2A routine prenatal ultrasound of a thirty-six-year old woman who was thirty weeks pregnant identified a right-sided pleural effusion in the fetus. The mother had gestational diabetes but was otherwise well. At thirty-four weeks’ gestation, the mother went into preterm labor at an outlying facility, and an ultrasound showed an increase in size of the fetal pleural effusion. Preterm labor was stopped, and the mother was transferred to a tertiary care center; maternal fetal medicine was consulted on arrival. An additional ultrasound showed a thoracic mass in the fetus, measuring 2.9 × 2.5 × 5.24 cm. Because of this mass and increasing pleural effusion, delivery was induced at thirty-five weeks. At birth, the neonate weighed 2.637 kg and was cyanotic with depressed respirations. She was stabilized and treated in the neonatal intensive care unit. Physical examination showed a well-appearing baby on continuous positive airway pressure, with a respiration rate of twenty-five breaths per minute. The patient had excellent breath sounds on the left, but she had diminished breath sounds on the right secondary to the pleural effusion. She had excellent chest rise without retraction. The initial chest radiograph showed a large right-sided effusion with a mediastinal shift. On the first day after delivery, a computed tomography (CT) scan of the chest showed a parenchymal mass abutting the anterior mediastinum, with associated right-sided pleural effusion. The mass, measuring 3.5 × 4.1 cm, was composed of soft tissue and fluid-filled cystic elements (Fig. 1). There were no calcifications in the mass, and it did not involve the heart or great vessels. Tracheal structures appeared normal with no involvement by the mass. A bone scan, performed to rule out metastatic disease, was normal. A magnetic resonance imaging study was considered but not performed. When the patient was seven days old, she underwent a posterolateral thoracotomy with resection of the mediastinal mass and removal of 40 mL of straw-colored fluid. The decision was made to proceed with primary resection instead of biopsy because the patient had no breath sounds on the right and had difficulty breathing. The mass extended both posteriorly and anteriorly, and it interdigitated between the fourth and fifth ribs and the fifth and sixth ribs. The mass was removed en bloc. There were also two additional lesions found, one on the pericardium and one along the posterior pleura; these lesions were also excised.
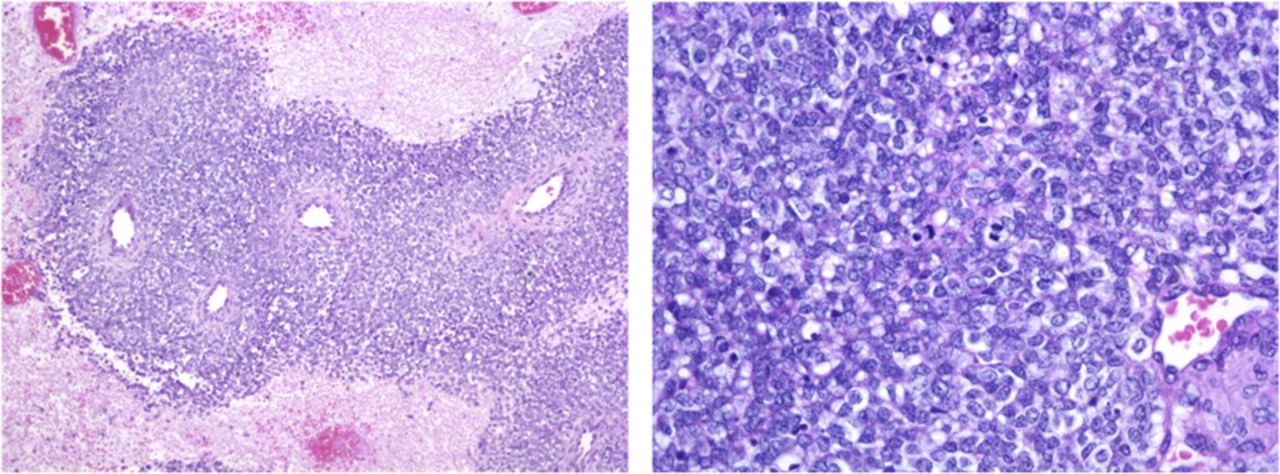
The mass weighed 23 g and measured 4.5 × 4 × 3 cm. Microscopic examination revealed a malignant neoplasm with a sheetlike proliferation of small to medium-sized blue cells. The cells had hyperchromatic nuclei, inconspicuous nucleoli, plentiful mitotic figures, and scant cytoplasm. There were multiple areas of tumor necrosis as well as focally associated dystrophic calcification. No rosettes were recognized. Tumor margins of the main mass were clear (Fig. 2). Immunohistochemical staining was positive for vimentin. Immunohistochemical stains for markers of muscle (desmin and myogenin) and lymphoma (cluster of differentiation [CD]45, terminal deoxynucleotidyl transferase [TdT]) differentiation were negative. Flow cytometry showed that the cells strongly expressed CD81, CD117, and CD99. Most of the cells were CD45 negative. Fluorescence in situ hybridization (FISH), looking for the gene fusion EWS-FL1 that results from t(11;22)(q24;q12) chromosomal translocation, was negative. The patient was diagnosed with primitive neuroectodermal tumor (PNET). The diagnosis of PNET was based on histological examination as well as flow cytometry, despite the negative result from the FISH test that was performed with an EWSR1 break-apart probe. According to the pathologist, the gene fusion EWS-FL1 is seen in 85% of patients with Ewing sarcoma, and variant fusions do exist. The patient recovered well from the surgical procedure and was discharged home on postoperative day eight. She was started on chemotherapy, which included cyclophosphamide, vincristine, and doxorubicin. The oncologist indicated that other than dose modification based on weight, there is no difference in chemotherapy treatment regime in neonates. There is a high level of concern for the long-term side effects of chemotherapy in neonatal patients. Our patient successfully finished the chemotherapy regime and had not had any recurrence two years postresection.
Proceed to Discussion >>Reference: Ghattas TN, Lucas G. Peripheral primitive neuroectodermal tumor of the chest wall in a neonate: a case report. JBJS Case Connect. 2013 Apr 24;3(2):e36.
Peripheral PNET and Ewing sarcoma are closely related, and the distinction between these two entities has become blurred. PNET/Ewing sarcoma is the most common tumor of the chest wall in children and adolescents. The literature shows that congenital and neonatal tumors are rare, accounting for only 2% of all childhood malignancies. Specifically, chest wall tumors are also a rare occurrence, comprising only 1.8% of all solid childhood tumors. There are multiple causes of these chest wall tumors, but the most common is the PNET/Ewing sarcoma/Askin tumor family, which comprises 71%. Askin et al. segregated a group of tumors of soft-tissue origin arising in the thoracopulmonary region, known as Askin tumors; 6.5% of primary tumors from these tumors originate in the chest wall. There is a clear predominance (75%) of chest wall tumors in girls. Initially PNET, Ewing sarcoma, and Askin tumors were considered to be separate entities, primarily because of tumor location. Masses that exhibited characteristics of small round blue cell tumors that arose in the soft tissue were labeled PNET, while those that arose in bone were labeled Ewing. Those found in the thoracopulmonary region were labeled Askin tumors. Askin had earlier segregated a group of thoracopulmonary soft-tissue tumors from a group of Ewing sarcomas that had neuroectodermal differentiation as well as other differences from PNET and Ewing tumors. In addition, similar histopathological findings, translocations, and responses to similar chemotherapy regimes further blurred the distinction between these entities. These disease processes are now considered a single entity called “malignant small round cell tumor” (MSRCT) and are grouped in the Ewing family of tumors. Most of these tumors have been translocations involving the EWS gene locus, which usually results in the EWS-FL1 fusion product due to the t(11;22)(q24;q12) translocation. The median age of presentation of MSRCT is fourteen to sixteen years. The patient described in this report actually was diagnosed prior to birth. To the best of our knowledge, only twenty-two cases of congenital PNET have been reported, none of which were diagnosed in utero, and only 14% occur in children under five years of age. These tumors are highly aggressive, and metastases are quite common at the time of diagnosis. The usual presentation for a child with a mediastinal or chest wall tumor is cough, chest pain, or a palpable mass. Infants and neonates may present with respiratory distress secondary to a pleural effusion or from an extensive intrathoracic mass. Evaluation begins with radiographs. CT scans should be obtained to further delineate the size of the tumor and any osseous involvement prior to surgical intervention. Bone scans are appropriate to rule out osseous metastases. Magnetic resonance imaging may be helpful to assess the extent of soft-tissue involvement. Treatment should be instituted as soon as possible. Chest wall tumors should be resected en bloc, with care taken to attain adequate tumor-free margins. Chemotherapy can either precede or follow surgical resection. Radiation therapy has been used in the past to help with local recurrence, but high incidences of recurrence with radiation therapy alone have lessened its use. Additionally, high doses of radiation therapy expose children to several adverse effects, such as growth retardation and secondary malignancy. Survival rates for patients with these tumors have improved dramatically because of advances in chemotherapy and surgical treatment. A retrospective review of twelve patients with malignant chest wall tumors (four Ewing, three PNET, one Askin, two rhabdomyosarcoma, and two neuroblastoma) reported that, at three years, seven (58%) were disease-free and ten (83%) had survived. All patients in that study had undergone surgical resection and chemotherapy. The average age at diagnosis was 8.9 years (range, six to sixteen years old). MSRCTs of the chest wall are rare in children and adolescents, but with advances in diagnostic and imaging tests, chemotherapy treatments, and surgical techniques, the prognosis for these children is improving.
Reference: Ghattas TN, Lucas G. Peripheral primitive neuroectodermal tumor of the chest wall in a neonate: a case report. JBJS Case Connect. 2013 Apr 24;3(2):e36.
Ewing sarcoma
Lymphoma
Primitive neuroectodermal tumor
Neuroblastoma
Wilms tumor